Ubinuclein 2 is essential for mouse development and functions in X chromosome inactivation
Asun Monfort, Giulio Di Minin, Sarah Sting, Charles Etienne Dumeau, Peter Scambler, Anton Wutz, Aleksandra Trifunovic, Aleksandra Trifunovic, Paula Cohen, Paula Cohen

TL;DR
Ubinuclein 2 is crucial for mouse development and helps in silencing one X chromosome in females.
Contribution
The study reveals a female-specific role of the HIRA complex in X chromosome inactivation and dosage compensation.
Findings
Ubn2 mutations cause embryonic lethality with a male-biased sex ratio.
HIRA complex is essential for initiating X chromosome inactivation by establishing H3K27me3 marks.
Combined Ubn1 and Ubn2 mutations lead to complete embryonic lethality and developmental arrest.
Abstract
The HIRA complex mediates deposition of histone H3.3 independent of replication. Its functions in gene regulation in mice remain to be fully understood. Here we analyze mutations of the HIRA complex genes Ubn1 and Ubn2. We observe that Ubn1 mutant mice of both sexes are viable and fertile. In contrast, mutation of Ubn2 causes embryonic lethality with variable penetrance and skewed sex ratio in favor of males. Combined Ubn1 and Ubn2 mutations cause embryonic lethality with complete penetrance, variable developmental arrest before turning, and reduced recovery of female embryos. Consistent with a female specific function of the HIRA complex, reanalysis of the Hira mutation during embryogenesis reveals that previously observed severe and mild phenotypic classes correspond to female and male sex. Mechanistically, we show that mutations of Ubn1, Ubn2, and Hira in mouse embryonic stem cells…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
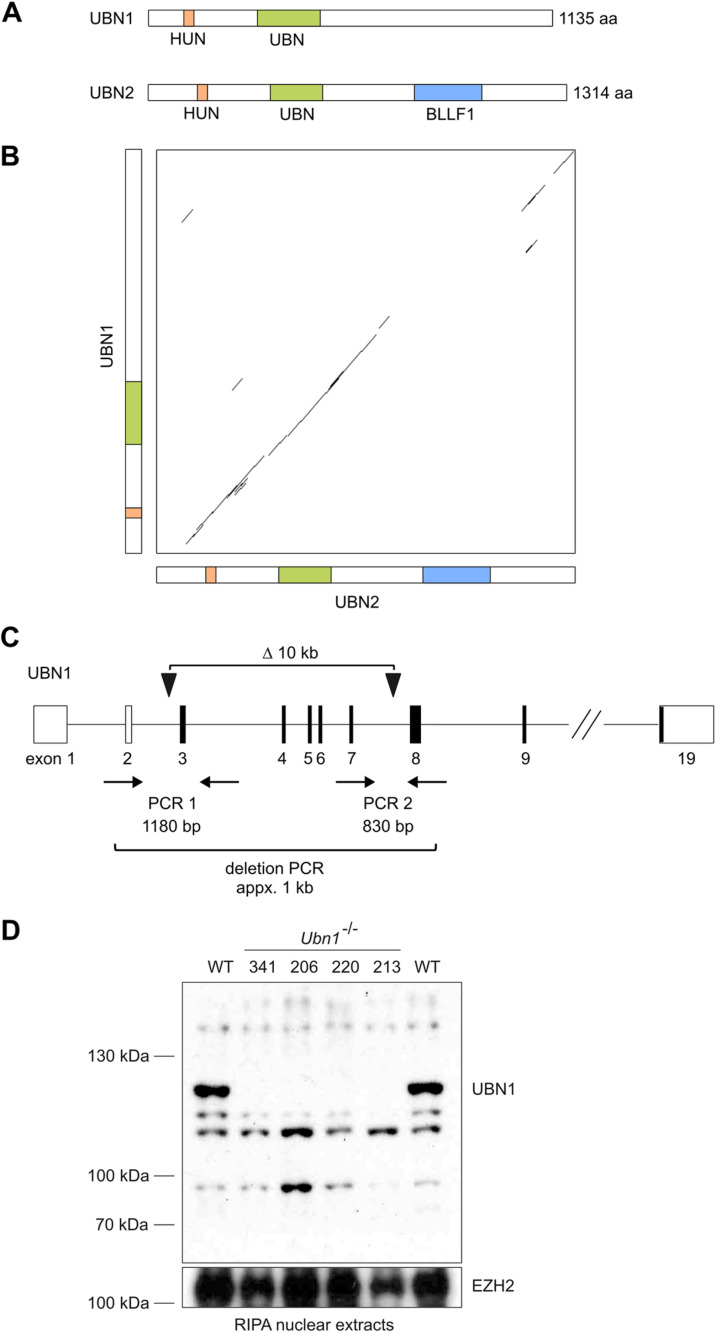 Fig 1
Fig 1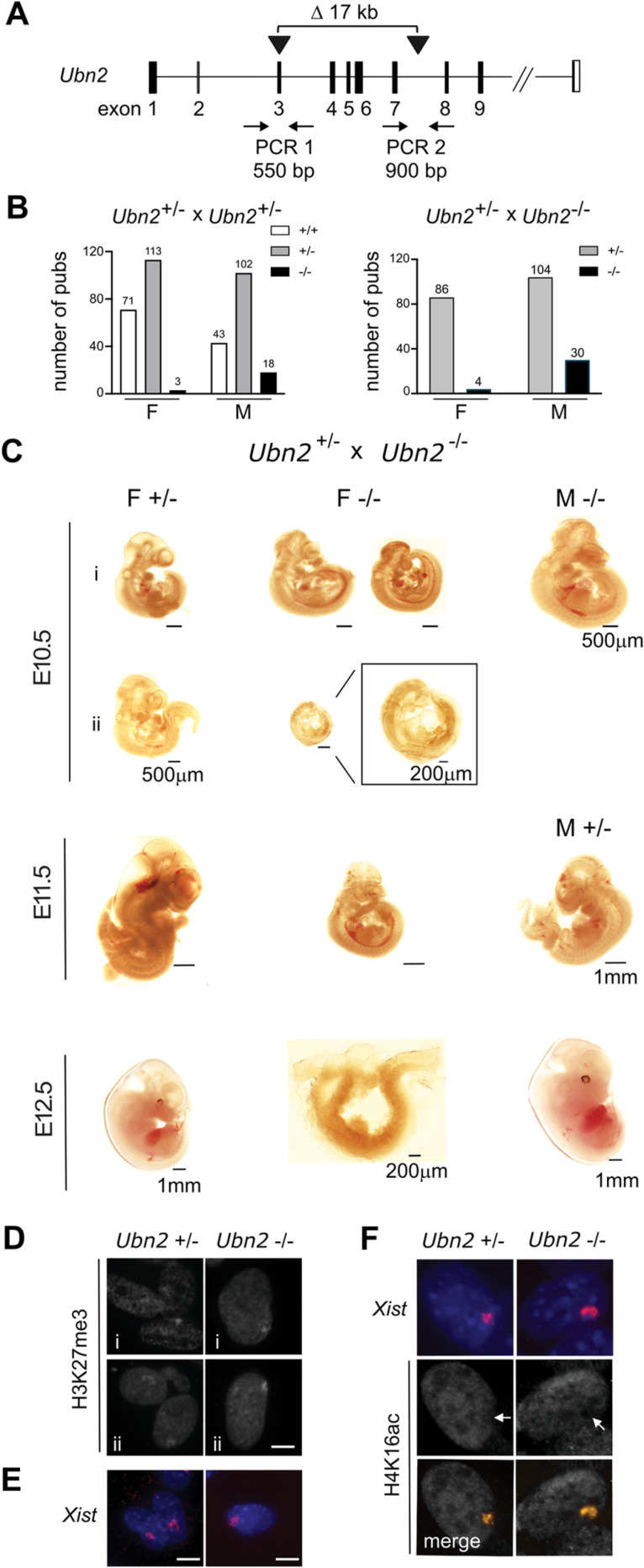 Fig 2
Fig 2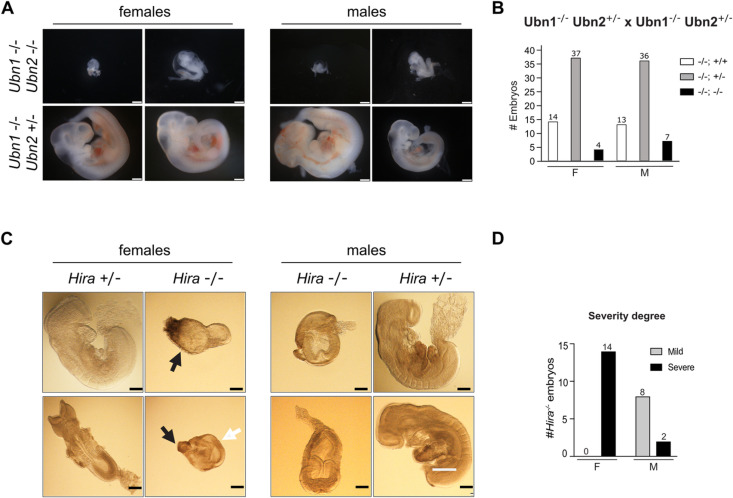 Fig 3
Fig 3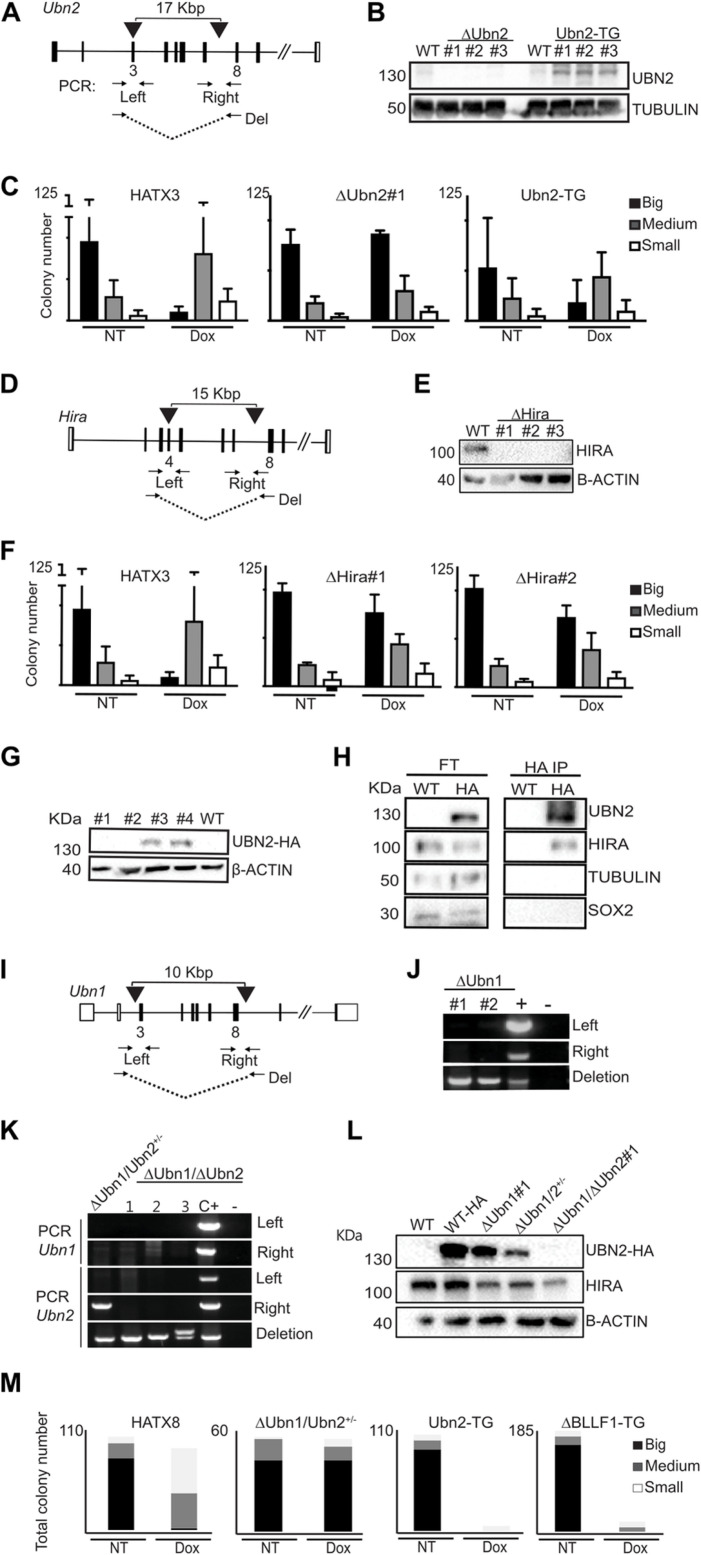 Fig 4
Fig 4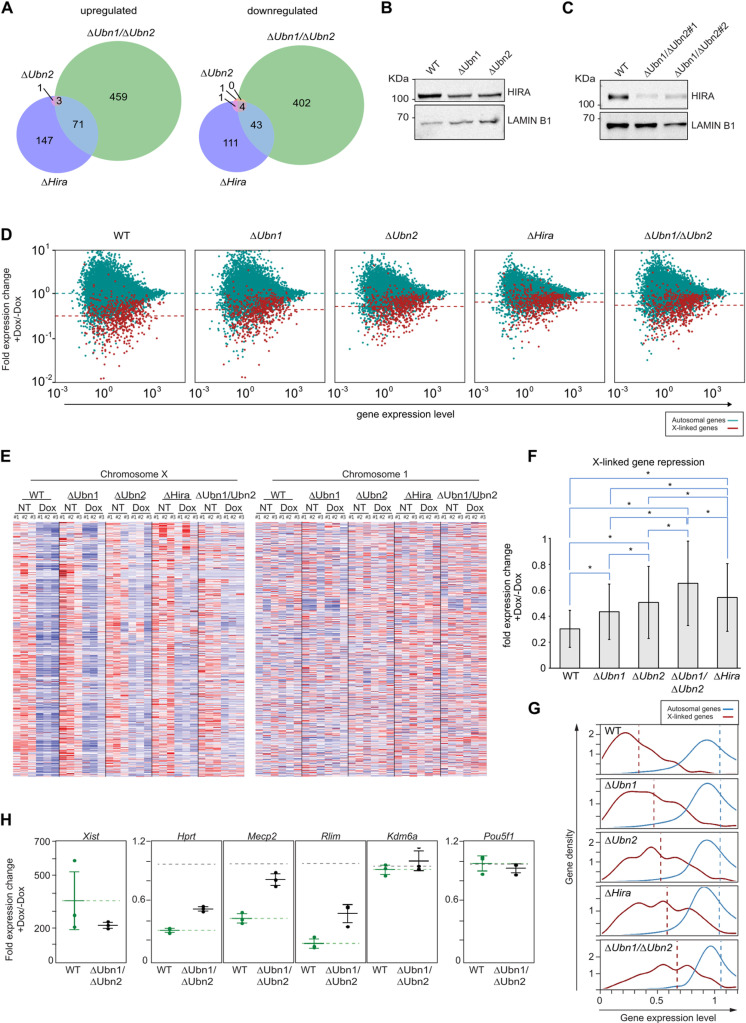 Fig 5
Fig 5 Fig 6
Fig 6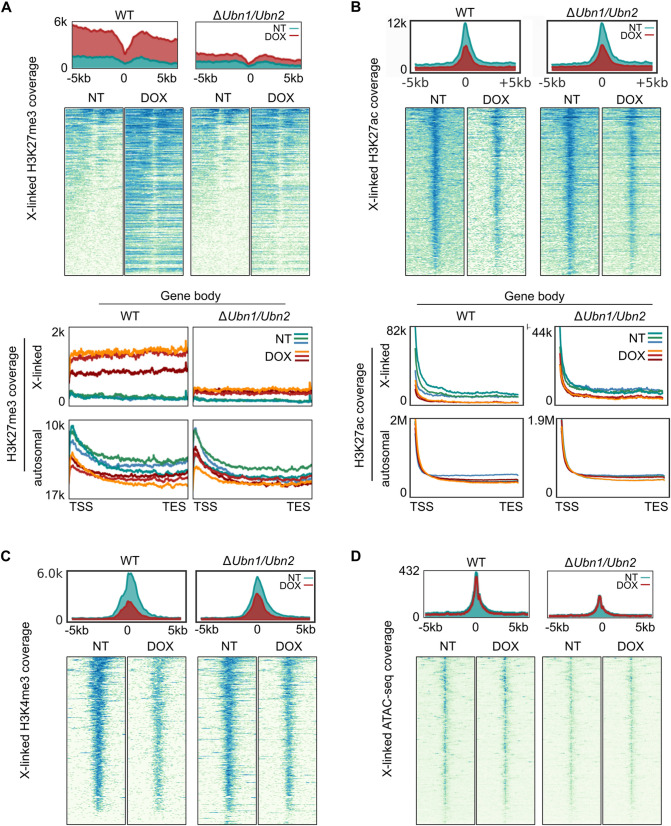 Fig 7
Fig 7- —http://dx.doi.org/10.13039/501100001711Schweizerischer Nationalfonds zur Förderung der Wissenschaftlichen Forschung
- —http://dx.doi.org/10.13039/501100001711Schweizerischer Nationalfonds zur Förderung der Wissenschaftlichen Forschung
- —http://dx.doi.org/10.13039/501100003006Eidgenössische Technische Hochschule Zürich
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsGenetic and Clinical Aspects of Sex Determination and Chromosomal Abnormalities · CRISPR and Genetic Engineering · Genetics and Neurodevelopmental Disorders
Introduction
Histone H3.3 is implicated in different processes including transcription [1], DNA repair [2], and genome stability [3]. Additionally, mutations in histone H3.3 occur in neurodevelopmental tumors [reviewed in 4]. In contrast to canonical histones H3.1 and H3.2, histone H3.3 deposition into chromatin is not restricted to S-phase. Histone regulator A (HIRA) and death domain associated protein (DAXX) complexes mediate histone H3.3 deposition by distinct mechanisms [reviewed in 5]. The latter forms a complex with alpha-thalassemia/mental retardation, X-linked (ATRX) integrating histone H3.3 into heterochromatin at telomeres, centromeres, and specific genomic elements which are associated with histone H3 tri-methyl lysine 9 (H3K9me3) [6,7]. Mutation of Atrx causes embryonic lethality in mice due to genome instability and trophoblast defects [8]. HIRA mediates histone H3.3 deposition in the transcription unit of active genes and at regulatory elements including superenhancers [9–11]. In addition, Hira is essential for reprogramming the zygotic genome after fertilization [12,13]. Mutation of Hira causes embryonic lethality shortly after implantation with mesodermal and patterning defects [14]. Surprisingly, mutation of Hira in embryonic stem cells (ESCs) has only minor effects on transcription [15] but Hira becomes essential when cells differentiate. Furthermore, HIRA and histone H3.3 are enriched at bivalent promoters and facilitate the establishment of histone H3 tri-methyl lysine 27 (H3K27me3) by Polycomb repressive complex 2 (PRC2) [16]. HIRA associates with the N-terminal NHRD domain of UBN1 through its WD repeats [17]. UBN1 binds histone H3.3 through an N-terminal Hpc2-related domain (HRD) [10] and has specificity for 3 amino acids in the core of histone H3.3 that differ from canonical histones H3.1 and H3.2 [4]. In mice, two genes, Ubn1 and Ubn2, encode proteins with homology to the Drosophila Yemanuclein and yeast Hpc2 genes [4]. Both share extensive similarity and have been shown to associate with HIRA [18,19], but their functions remain to be determined in mice.
We have previously identified Hira and Ubn2 as candidate genes for silencing factors in X chromosome inactivation (XCI) [20]. XCI is the mechanism for dosage compensation between males and females in mammals [reviewed in 21]. The non-coding Xist RNA is transcribed from and associates specifically with the inactive X chromosome (Xi) leading to silencing of X-linked genes [22,23]. Spreading of Xist over the Xi forms a repressive compartment that is depleted of RNA polymerase II, splicing and transcription factors, as well as acetylated histone H3 and H4 [24]. Subsequently, PRC1 and PRC2 are recruited and establish mono-ubiquitination of histone H2A lysine 119 (H2AK119ub) and H3K27me3, respectively [25,26]. Recruitment of Polycomb complexes is facilitated by Xist repeats B and C, and a pathway involving heterogeneous ribonucleoprotein K (hnRNPK) [25,27–29]. Notably, the initial repressive compartment forms over regions that are devoid of genes and genes only associate with the Xist domain upon silencing [24]. Binding of the split ends protein SPEN to Xist repeat A is required for X-linked gene silencing by an HDAC3 dependent mechanism [20,27,30,31]. In somatic cells, the Xi forms a stable heterochromatic structure and gene repression becomes independent of Xist [reviewed in 21].
We previously used an inducible Xist expression system in mouse haploid ESCs for screening of silencing factors in XCI [20]. Induction of Xist led to rapid silencing of the single X chromosome and caused cell death. Strong selection for Ubn2 and Hira mutations in surviving cells after Xist induction indicated a potential role in XCI. Specifically, we hypothesize that Hira and Ubn2 might function in Polycomb recruitment and gene silencing. Here, we engineer Ubn1 and Ubn2 mutations in mice and ESCs to characterize their role in embryonic development and XCI.
Results
Ubn1 mutant mice are viable and fertile
Ubn1 and Ubn2 are the mammalian ortholog and paralog of the Drosophila Yemanuclein and yeast Hpc2 genes, respectively [1,4,32] (Fig 1A and 1B). In mouse zygotes, RNA interference mediated depletion of Ubn1 has been reported to abrogate preimplantation development [33]. We previously engineered an approximately 10kb deletion of Ubn1 exons 1–7 encoding the N-terminal NHRD and HRD domains (also encompassing the HUN domain), which are required for interaction with HIRA and histone H3.3 [10,17,32] (Fig 1C) in mice. For this we performed zygote electroporation of a pair of Cas12a/Cpf1 nuclease gRNA complexes and obtained several founder mice in the C57BL/6 background [34]. From crosses we obtained homozygous Ubn1 mutant offspring of both sexes that appeared healthy and fertile. We subsequently established a homozygous Ubn1 mutant mouse colony and confirmed the absence of UBN1 protein in liver nuclear extracts by Western analysis (Fig 1D). Over the past 5 years no overt phenotypic alteration was observed and average litter sizes of 5.31 + /- 2.3 and 5.93 + /- 2.2 were calculated for Ubn1^-/-^ and control wildtype C57BL/6 mice, respectively (n > 160 litters) suggesting comparable breeding success. Both Ubn1^-/-^ and wildtype colonies showed a balanced sex ratio. We conclude that Ubn1 is not essential in mice.
Ubn1 mutation in mice.(A) Scheme showing the domain structure, and (B) Dot-plot showing protein similarity (Dotmatcher, windowsize = 30, threshold = 40) of the mouse UBN1 and UBN2 proteins. (C) Scheme showing the strategy for engineering a deletion in Ubn1 in mice. (D) Western analysis of UBN1 protein in liver nuclear extracts. EZH2 was used to control for loading.
Embryonic lethality of the Ubn2 mutation with incomplete penetrance and male biased survival
The absence of a developmental phenotype in Ubn1 mutant mice indicated possible compensation by Ubn2. We therefore engineered a 17kb deletion of Ubn2 exons 3–7 encoding the N-terminal HUN domain [32] that is predicted to introduce a frameshift (Fig 2A). We obtained several founders with deletions and inversions in Ubn2 from pronuclear microinjection of a pair of Cas9 nuclease-gRNA complexes into C57BL/6 zygotes. Crosses of heterozygous Ubn2^+/-^ mice yielded several homozygous Ubn2^-/-^ mutant males at submendelian frequency that were healthy and fertile (Fig 2B). However, initially no females were found at weaning. A female specific lethality was further substantiated by crossing homozygous Ubn2^-/-^ males with heterozygous Ubn2^+/-^ females (Fig 2B). Taken together, all crosses yielded only 7 Ubn2^-/-^ females against a total of 48 Ubn2^-/-^ males (S1 Table). Therefore, homozygous Ubn2 mutant females in the C57BL/6 background were rarely obtained and did not produce offspring when mated with either mutant or wild type males. To determine at which developmental stage Ubn2^-/-^ embryos were lost, we analyzed embryos at embryonic days (E) 10.5, E11.5, and E12.5. We recovered mutant embryos of both sexes and observed a large variability of embryonic delay (Figs 2C, and S1). Some male homozygous mutant embryos appeared comparable to control heterozygous embryos consistent with survival of Ubn2^-/-^ males at submendelian frequency. Homozygous Ubn2^-/-^ mutant female embryos could be recovered at all stages but showed a developmental delay with frequent arrest before E10.5 (Fig 2C). We established fibroblasts from two E11.5 female Ubn2^-/-^ embryos. Immunofluorescence and RNA FISH analysis revealed H3K27me3 foci and Xist clusters comparable to controls suggesting that XCI had been initiated (Fig 2D and 2E). Furthermore, H4K16ac was depleted from the Xist domains indicating that Xist formed a repressive compartment (Fig 2F). We conclude that the Ubn2 mutation causes embryonic lethality with incomplete penetrance and biased survival of males in the C57BL/6 background.
Ubn2 mutation causes a female specific lethality with high penetrance in C57BL/6 mice.(A) Scheme showing the strategy for engineering a deletion in Ubn2 in mice. (B) Male (M) and female (F) offspring from crosses of mice as indicated. Numbers above columns represent the number of mice. (C) Female specific phenotype of Ubn2-/- embryos at E10.5, 11.5, and 12.5. i) and ii) represent two independent litters. (D) H3K27me3 immunofluorescence, (E) Xist RNA FISH, and (F) Combined Xist RNA FISH and H4ac immunofluorescence of E11.5 Ubn2-/- mouse embryonic fibroblasts (MEFs). i, and ii represent two independent cultures. Scale bar, 5 µm.
Combined Ubn1 and Ubn2 mutations cause embryonic lethality with complete penetrance
To investigate if Ubn1 mediated the survival of Ubn2^-/-^ mice, we introduced the Ubn2 mutation into the Ubn1 homozygous mutant C57BL/6 background by crossing. We obtained mice with heterozygous Ubn2 and homozygous Ubn1 mutations of both sexes. However, we did not obtain double homozygous mutant Ubn1^-/-^ Ubn2^-/-^ mice suggesting embryonic lethality. We next analyzed embryos from crosses of Ubn1^-/-^ Ubn2^+/-^ mice at E10.5. We recovered several homozygous double-mutant Ubn1^-/-^ Ubn2^-/-^ embryos that showed an embryonic delay and arrest before turning (Fig 3A). We performed Chi-Square tests and observed a high deviation from expectations for double homozygous embryos that, however, did not reach statistical significance (S2 Table). We also noted an elevated frequency of Ubn2 heterozygous and Ubn1 homozygous mutants that might indicate potential maternal contamination. All genotypes approach Mendelian frequencies if we consider that maternal contamination occurred at some poorly developed Ubn1^-/-^ Ubn2^-/-^ implantation sites. None of the Ubn1^-/-^ Ubn2^-/-^ embryos had initiated turning demonstrating a severe developmental delay. The high variability of embryonic delay among Ubn1^-/-^ Ubn2^-/-^ E10.5 embryos was striking with a 2-fold higher recovery of Ubn1^-/-^ Ubn2^-/-^ male compared to female embryos (Fig 3B).
Early embryonic lethality of the Ubn1 and Ubn2 double and Hira mutations in mice.(A) Representative images of male and female Ubn1-/- Ubn2-/- and control Ubn1-/- Ubn2+/- E10.5 embryos. Scale bar, 500 µm. (B) Genotype distribution of embryos obtained from crossing Ubn1-/- Ubn2+/- mice. (C) Images of male (M) and female (F) Hira-/- embryos at E8.5. Embryonic (white arrows) and extraembryonic tissues (black arrows) are indicated. Scale bar, 200 µm. (D) Phenotypic classes, mild and severe, of male (M) and female (F) Hira-/- E8.5 embryos. Number of embryos is indicated on top of columns.
Two phenotypic classes of the Hira mutation correlate with sex of the embryos
Consistent with our observation in Ubn1^-/-^ Ubn2^-/-^ mice, embryonic lethality after gastrulation has previously been observed for the Hira mutation in inbred C57BL/6 mice. In the outbred CD1 background embryos could be recovered until E10.5, whereby two classes of phenotypes were reported, namely a mildly affected and a severely affected class [14]. Reduced recovery of female Ubn2^-/-^ mice and Ubn1^-/-^ Ubn2^-/-^ embryos suggested the possibility that the two classes of the Hira^-/-^ embryos were also correlated with sex. We therefore established crosses of heterozygous Hira mutant mice in the CD1 background. Among 146 embryos (73 females and males) were 14 and 10 Hira^-/-^ females and males, respectively. All Hira^-/-^ embryos were markedly smaller than wildtype or heterozygous littermates (Fig 3C). Female Hira^-/-^ embryos showed a more severe developmental delay as judged by size and earlier arrest than male Hira^-/-^ embryos (Fig 3D). When assigned into two phenotypic classes, all female embryos clearly associated with the severe phenotype. In contrast, the majority of male homozygous mutant embryos developed further and displayed milder phenotypes. Hira^-/-^ embryos of both classes arrested before turning consistent with previous observations [14]. We conclude that sex of the embryo correlates with two phenotypic classes of the Hira mutation in the CD1 background.
Hira and Ubn2 complexes contribute to Xist function in mouse ESCs
Our finding that mutations in Ubn2 and Hira affected female more than male development were consistent with previous identification of both genes in our screen for silencing factors in XCI [20]. To study their role in XCI further, we engineered mutations in ESCs that contain an inducible Xist expression system. A tetracycline-inducible promoter (tetOP) was inserted at the Xist transcription start site and a tetracycline-regulated transactivator targeted into the ubiquitously expressed ROSA 26 locus on chromosome 6 [20] (S2A Fig). Induction of Xist with doxycycline causes cell death due to X-linked gene silencing, which provides a readout for Xist function. Initially, these cells were derived as haploid ESCs [20], but became diploid over time and, hence, possess two X chromosomes from which Xist can be induced. We engineered Ubn2 mutations in two independent ESC lines, HATX3 and HATX8 [20], using Cas9 nucleases with either a single gRNA to generate frame shift mutations in exon 3, or a pair of gRNAs to make a deletion from exon 3 to intron 7 (Fig 4A). We obtained several ΔUbn2 ESC lines that proliferated normally and maintained a typical colony morphology suggesting that self-renewal of ESCs was not impaired. UBN2 protein was undetectable by Western analysis (Figs 4B and S2B–S2D). To assess the effect of Ubn2 mutations on Xist function, we measured the survival of ESCs after Xist induction using a clonal assay, where the number and size of colonies in the presence relative to the absence of doxycycline was recorded (S2E Fig). In the parental ESCs Xist induction drastically reduced colony size and number, whereas three independent ΔUbn2 ESC clones showed greater survival after Xist induction than the parental WT ESCs (Figs 4C, S2F and S2G). Importantly, complementation of the Ubn2 mutation by stable integration of a piggyBac vector expressing a Ubn2 cDNA transgene restored Xist function (Figs 4B, 4C, S2F and S2H). Similarly, we engineered mutations in Hira (Figs 4D and S2I and S2J) and confirmed the absence of HIRA protein in ΔHira ESCs by Western analysis (Fig 4E). Independent ΔHira ESC clones showed increased cell survival after Xist induction compared to wildtype controls (Figs 4F and S2K). To confirm formation of complexes containing UBN2 and HIRA in our ESC system, we introduced a hemagglutinin-tag (HA) into a C-terminal position of the endogenous Ubn2 gene locus using Cas9 nuclease mediated homology directed repair. HA-tagged UBN2 protein in Ubn2-HA ESCs was detected by Western analysis (Fig 4G). Western analysis of HA-immunoprecipitates of nuclear extracts of Ubn2-HA ESCs showed that HIRA protein was present confirming that HIRA interacts with UBN2 (Fig 4H). To obtain additional support, we performed preliminary characterization of this complex by comparative proteomics of SILAC-labelled Ubn2-HA ESCs and control untagged ESCs. We observed enrichment of HIRA, UBN1, and CABIN1 in UBN2-HA over control precipitates (S3 Table), which is further consistent with a recent report [10]. Taken together our experiments showed that UBN2 and HIRA form complexes in ESCs and contribute to Xist function.
Ubn2 and Hira are required for XCI.(A) Scheme showing the positions of gRNAs (black arrow heads) and PCR primers (arrows) for engineering deletions in ΔUbn2 clones #1 and #2, and a frame-shift in exon 3 in ΔUbn2#3. (B) Western analysis of UBN2 protein in control WT, ΔUbn2, and transgenically (TG) rescued Ubn2-TG ESCs is shown. TUBULIN used as loading control. (C) Representative Xist survival assay for control HATX3, ΔUbn2, and rescued Ubn2-TG ESCs as indicated. For each cell line colony numbers binned in 3 size classes are shown without (NT) or with (Dox) Xist induction (n = 3). (D) Strategy for disrupting Hira as in (A). Deletions were introduced into ΔHira clones #1 and #2, and a frame-shift in exon 4 in ΔHira#3. (E) Western analysis of HIRA protein in control WT, and ΔHira ESCs. β-ACTIN used as loading control. (F) Representative Xist survival assay for control HATX3, ΔHira#1 and ΔHira#2 ESC clones without (NT) or with (Dox) Xist induction (n = 3) is shown as in (C). (G) Western analysis showing UBN2-HA protein expression in Ubn2-HA ESC clones #3 and #4 using HA antisera. β-ACTIN used as loading control. (H) Western analysis detecting HIRA in HA-immunoprecipitates from nuclear extracts of Ubn2-HA ESCs using antisera as indicated. FT; flow through. (I) Strategy for disrupting Ubn1 in Ubn2-HA ESCs as in (A). (J) Genomic PCR identifying deletions in ΔUbn1 ESC clones #1 and #2. + ; positive control heterozygous for the ΔUbn1 deletion. (K) Genomic PCR showing deletions in four independent ΔUbn1/ΔUbn2 double mutant ESC clones. A band is observed for the right flanking PCR in ΔUbn1/ΔUbn2+/- ESCs indicating a partial deletion. C + ; positive control heterozygous for the ΔUbn1 and ΔUbn2 deletions. (L) Western analysis of UBN2-HA and HIRA in WT, ΔUbn1, and ΔUbn1/ΔUbn2 ESCs. ΔUbn1/ΔUbn2+/- ESCs show a reduced expression of truncated UBN2-HA and appear to be functionally comparable to ΔUbn1/ΔUbn2 ESC clones. β-ACTIN used as loading control. (M) Representative Xist survival assay for ΔUbn1/ΔUbn2+/- ESCs rescued with a WT UBN2 and a ΔBLLF1-UBN2 cDNA transgene.
To further investigate if Ubn1 also contributes to Xist function, we engineered an Ubn1 deletion in an Ubn2-HA ESC line that was homozygous for the HA-tagged Ubn2 allele (Fig 4G and 4I). We also generated a Ubn1 and Ubn2 double mutation by subsequently introducing a deletion into Ubn2. We obtained several Ubn2-HA ΔUbn1 mutant ESC clones (Fig 4J) and recovered one Ubn2-HA ΔUbn1/Ubn2^+/-^ (with a heterozygous Ubn2 mutation, where one allele maintains sequences on the right flank, Fig 4K) and three homozygous Ubn2-HA ΔUbn1/ΔUbn2 ESC clones (Fig 4K). Western analysis confirmed the reduction and absence of the Ubn2-HA protein in Ubn2-HA ΔUbn1/Ubn2^+/-^ and Ubn2-HA ΔUbn1/ΔUbn2 ESC clones, respectively (Fig 4L). Ubn2-HA ΔUbn1 ESCs showed increased survival after Xist induction, when compared to the parental WT cells (S3A Fig). Ubn2-HA ΔUbn1/ΔUbn2 double mutant ESCs showed higher survival after Xist induction than ΔUbn1 cells and comparable to the survival of ΔUbn2 ESCs (S3A, S3B Fig). Thus, albeit both Ubinucleins contribute to Xist function, Ubn2 has a predominant role. UBN2 possesses a BLLF1 domain of unknown function that is absent in UBN1 (Figs 1A, 1B, and S3C). To investigate a role of the BLLF1 domain for Xist function, we introduced piggyBac vectors for stable expression of WT and ΔBLLF1 Ubn2 cDNAs into ΔUbn2 and Ubn2-HA ΔUbn1/Ubn2^+/-^ ESCs. Both transgenes restored Xist function with comparable efficiency suggesting that the BLLF1 domain is not essential for Xist function (Figs 4M, and S3B). Transgenic rescue of Xist function confirmed that the effects on Xist were specific and caused by the Ubn2 mutation.
Xist mediated X-linked gene repression is impaired by Hira and Ubn2 mutations
We next investigated the effect of our mutations on the transcriptome of ESCs and the repression of X-linked genes by Xist. For this, we performed RNA-Seq for three biological replicates of WT, ΔUbn1, ΔUbn2, ΔUbn1/ΔUbn2, and ΔHira ESCs either without or after 48 hours of Xist induction. RNA-Seq profiles for the different clones confirmed the expected absence of transcripts overlapping the deleted exons (S4A and S4B Fig). Analysis of gene expression differences to WT controls without Xist induction using DESeq2 [35] revealed that only 5 and 13 genes out of 15329 with nonzero total read count were significantly differentially expressed (adj.p-value < 0.05) in Ubn1 and Ubn2 mutant ESCs, respectively (S4 Table). In ΔUbn1/ΔUbn2 and ΔHira ESCs, 1745 and 703 genes were significantly differentially regulated, respectively (S4C Fig, and S4 Table). Of these, 222 and 159 genes were 2-fold upregulated and downregulated in Hira mutant ESCs, respectively. 533 and 449 genes were 2-fold upregulated or downregulated in ΔUbn1/ΔUbn2 ESCs. Notably, there was little overlap of differentially expressed genes between ESCs carrying different mutations (Fig 5A). Some of the downregulation might be explained by loss of the second X chromosome in ΔUbn1/ΔUbn2 ESCs (S3D Fig), but also among upregulated genes only a minority overlaps between ΔHira and ΔUbn1/ΔUbn2 ESCs, which suggests biological differences. Known regulators of pluripotency remained unaffected by any of the mutations (S4 Table), which was further consistent with robust ESC self-renewal. Hira, Ubn1, and Ubn2 transcripts remained unaffected by any mutation in the respective other genes indicating that transcriptional compensatory upregulation does not occur in ESCs (S4 Table). In contrast, Ubn1 and Ubn2 mutations reduced HIRA protein level in mice (Fig 5B), and in ΔUbn1/ΔUbn2 ESCs a drastic reduction of HIRA protein was evident by Western analysis (Fig 5C). This observation is consistent with previous reports of interdependence of HIRA complex proteins [2,10].
*Ubinucleins and Hira are required for efficient gene repression by Xist.RNA expression analysis of WT, ΔUbn1, ΔUbn2, ΔHira, and ΔUbn1/ΔUbn2 ESCs. (A) Venn diagrams showing number of 2-fold or more up- (left) and down-regulated (right) genes in biological triplicates of ΔUbn2, ΔHira and ΔUbn1/ ΔUbn2 relative to WT ESCs with an adjusted p-value cutoff of 0.05. (B) Western analysis of HIRA in ΔUbn1 and ΔUbn2 mice. (C) Western analysis of HIRA in ΔUbn1/ ΔUbn2 cells. Lamin B1 was used as loading control. (D) Fold expression change of autosomal (green) and X-linked genes (red) after 48h of Dox treatment of ESCs with indicated genotypes versus their expression level in control condition. Dotted lines indicate the median fold change of all autosomal (green) and X-linked genes (red). (E) Heatmap of normalized and z-transformed expression of genes in order of chromosomal positon is shown for ESCs of genotypes indicated, without (NT) and after 48h of Xist induction (Dox). Chromosome 1 is shown as autosomal control. Red and blue color represent lower and higher expression from mean expression of the gene, respectively. White color indicates mean expression. (F) Bars graph of fold change expression of X-linked genes after 48h of Xist induction relative to uninduced controls. Genes with a fold-change of less than 0.6 in WT were selected and mean and standard deviation (errorbars) of fold change are shown for all genotypes after induction. ; p-value < 0.001 from a paired 2-sided t-test between groups at end of blue lines. (G) Efficiency of X-linked gene repression by Xist: Histograms (bins 0.1) show the distribution of gene numbers with indicated fold expression change after 48h of Xist induction (Dox) in combined replicates of WT and mutant ESCs. Autosomal (blue) and X-linked genes (red) are shown separately and their median fold expression change is indicated by vertical dotted lines. (H) Fold expression change for the X-linked Xist, Hprt, Mecp2, Rlim, and the escape genes Kdm6a, after 48h of treatment with Dox in WT and ΔUbn1/ ΔUbn2 ESCs. Pou5f1 is showed as an autosomal control. Dots represent replicates, mean and standard deviation are indicated by wide and narrow horizontal bars, respectively.
Next, we studied the effect of Xist induction on X-linked gene expression in the different mutant ESCs. In RNA-Seq datasets from ESCs that were treated with Dox for 48h, Xist was strongly up-regulated. Conversely, we observed a reduction in X-linked gene expression (Figs 5D, 5E, and S4D, S4E). Although a repression of X-linked genes was observed in all mutant ESCs, the strength of repression was markedly reduced compared to WT ESCs (Fig 5D–5G). In addition, we noted that Xist function was increasingly impaired along the genotype order ΔUbn1, ΔUbn2, ΔHira, and ΔUbn1/Ubn2. Loss of silencing efficiency of Xist was observed in a shift in the average fold expression change obtained over the three independent replicates for every mutation (Figs 5D, 5F, 5G and S4D) as well as when X-linked gene expression was represented in a heatmap for genes along the X-chromosome (Fig 5E). The observation that the Hira mutation and the Ubn1 and Ubn2 double mutation had the strongest impact on gene repression by Xist suggests redundant roles of Ubn1 and Ubn2 in ESCs (Fig 5D–5G). The strength of repression by Xist showed some variability over the X chromosome but was not correlated with the distance from the Xist locus (S4E Fig). Genes that escape XCI and autosomal genes were not affected by Xist induction (Fig 5E, 5H). Our results show that Ubn2 and Hira are required for efficient Xist-mediated gene silencing, whilst mutation of Ubn1 affected Xist only weakly and in combination with mutation of Ubn2.
Xist forms a repressive compartment in the absence of Ubn1 and Ubn2
Next, we investigated which steps of XCI depend on Ubinucleins. We performed RNA-FISH to examine Xist expression in WT, and ΔUbn1/ΔUbn2 mutant ESCs (Fig 6). Xist clusters were observed after induction with doxycycline and their shape and size were comparable suggesting that Xist localization was not abrogated by the loss of Ubinucleins (Fig 6A). A slight decrease in the number of cells with Xist clusters was observed in ΔUbn1/ΔUbn2 mutant compared to control ESCs (Fig 6B). To test if HIRA or UBN2 were enriched on the X chromosome after Xist induction, we performed immunofluorescence. We observed a diffusely punctate nuclear pattern for HIRA without a focal enrichment over the Xist domain (S5A Fig). Similarly, we did not detect a focus of UBN2-HA using an HA antiserum after induction of Xist in UBN2-HA ESCs (S5A Fig). Taken together our observations indicate that Xist does not recruit the HIRA complex suggesting that it might act locally on chromatin where it is already bound as has been previously suggested for hnRNPK, and HDAC3 [31].
*Formation of a repressive compartment by Xist in Ubinuclein, and Hira mutant ESCs.(A) Xist RNA FISH in WT and ΔUbn1/ΔUbn2 cells and (B) quantification of Xist RNA localization patterns after 24h of Xist induction (cells counted WT: 372, Ubn1/2 KO #1: 402, Ubn1/2 KO #2: 426). (C) Immunofluorescence staining for H3K27me3 in WT and ΔUbn1/ΔUbn2 and (D) quantification of foci formation after 24h of Xist induction (cells counted WT: 180, Ubn1/2 KO #1: 155, Ubn1/2 KO #2: 294). (E) Immunofluorescence staining for H2AK119Ub in WT and ΔUbn1/ΔUbn2 and (F) quantification of foci formation after 24h of Xist induction (cells counted WT: 221, Ubn1/2 KO #1: 118, Ubn1/2 KO #2: 189). (G) Double immunofluorescence staining in ΔUbn1/ΔUbn2 double mutant ESCs after 24h of Xist induction using H4K16ac or H3K27ac with EZH2 and JARID2 antisera, respectively. Scalebar; 5 µm. (H) Quantification of JARID2 and EZH2 foci in ΔUbn1/ΔUbn2#1 ESCs after 24h of Xist induction. ns; P > 0.05, *; P ≤ 0.05, **; P ≤ 0.01, **; P ≤ 0.001 (cells counted WT: 319, Ubn1/2 KO #1: 312, Ubn1/2 KO #2: 309).
We next investigated if Xist could form a repressive compartment. For this we performed combined immunofluorescence and Xist RNA FISH experiments using antibodies specific for H3K27me3 and H2AK119ub1, as well as JARID2 and EZH2 (Figs 6C, 6E, 6G, and S5B–S5H). We observed focal staining overlapping with Xist clusters for all antisera demonstrating that a repressive compartment and associated chromatin modifications can be established by Xist in ΔUbn1, ΔUbn2, ΔUbn1/ΔUbn2, and ΔHira ESCs. The percentage of nuclei showing focal domains of H3K27me3 and H2AK119ub1 was significantly reduced in ΔUbn1/ΔUbn2 ESCs (Fig 6D, 6F), whereas the reduction in focal JARID2 and Ezh2 remained below statistical significance (Fig 6H). Taken together our data show that a domain of repressed chromatin was established by Xist in the absence of Ubinucleins or Hira.
Ubinucleins are required for switching H3K27ac to H3K27me3 over X-linked genes
Xist forms a repressive compartment over non-genic regions of the X chromosome that has been separated from gene repression [24,36]. Gene repression requires Xist repeat A and Spen dependent pathways [20,30,31]. Previous studies have shown that Ubinucleins and Hira mediate histone H3.3 deposition and establishment of H3K27me3 over promoters and regulatory regions in ESCs [10,16]. To investigate effects on X-linked genes at higher resolution, we performed native chromatin immunoprecipitation sequencing (nChIP-Seq) with an H3K27me3 specific antiserum. For this we analyzed three independent WT and ΔUbn1/ΔUbn2 ESC clones without or after 48 hours of Xist induction. Before Xist induction promoters of low and medium expressed genes had on average higher H3K27me3 signals than highly expressed genes in both WT and ΔUbn1/ΔUbn2 ESCs as expected (S6A–S6C Fig). After Xist induction H3K27me3 increased for all groups of X-linked promoters and gene bodies (Figs 7A, and S6A–S6C). However, the increase was substantially smaller in ΔUbn1/ΔUbn2 than WT ESCs, whereby the largest difference was seen for highly expressed genes (Figs 7A, and S6A–S6C). Plotting of fold-change of H3K27me3 against fold-change in gene expression showed that genes, whose repression by Xist is affected in ΔUbn1/ΔUbn2 ESCs, are also failing to acquire H3K27me3 at their TSS and gene body (S6B Fig). We observed a bimodal distribution of H3K27me3 around the transcription start site (TSS) of highly or moderately expressed X-linked genes, whereas low expressed genes showed a uniform increase (S6A, S6C Fig). To understand if H3K27me3 establishment over regulatory elements was also affected by mutations in Ubn1 and Ubn2, we analyzed 5kb intervals around the center of previously described elements. We observed modest increases in H3K27me3 after Xist induction over X-linked strong EZH2 sites [26] and CpG islands in WT ESCs that was slightly reduced in ΔUbn1/ΔUbn2 ESCs (S6D Fig). Over X-linked moderate EZH2 sites [26], X-linked Formaldehyde-Assisted Isolated Regulatory Elements (FAIRE), CTCF sites [37], predicted enhancers [38], and the transcription start sites (TSS) of RefSeq genes Xist induction increased H3K27me3 strongly in WT ESC but only weakly in ΔUbn1/ΔUbn2 ESCs (S6D Fig). Autosomal nChIP-Seq read coverage was similar for all ESC clones and independent of Xist induction and genotype (Figs 7A and S6C). We conclude that Xist induction strongly increased H3K27me3 specifically over X-linked genes and regulatory elements in WT ESCs but only weakly in ΔUbn1/ΔUbn2 ESCs.
Ubinucleins are required for H3K27ac to H3K27me3 switch during initiation of XCI.Profiles and heatmaps showing cumulative (A) H3K27me3 ChIP coverage, (B) H3K27ac CUT&RUN signals, (C) H3K4me3 CUT&RUN coverage, and (D) ATACseq coverage over X-linked highly expressed genes for three independent replicates (AB) or a representative experiment (CD) of WT and ΔUbn1/ΔUbn2 ESC clones after 48h of Xist induction (DOX) and without (NT). Profiles of 5kb around the TSS are shown. (AB) Profiles over the scaled transcription units of X-lined and autosomal highly expressed genes are shown for each of the three biological replicates (lower panels).
To investigate whether persisting active chromatin modifications at promoters might prevent establishment of H3K27me3 at X-linked genes after Xist induction in ΔUbn1/ΔUbn2 ESCs, we performed CUT&RUN analysis using antibodies specific for H3K27ac and H3K4me3 (Fig 7B, 7C). Defined H3K27ac peaks were observed over the TSS of active genes in WT and ΔUbn1/ΔUbn2 ESCs as expected. After Xist induction H3K27ac was markedly reduced at X-linked but not autosomal genes in WT and ΔUbn1/ΔUbn2 ESCs demonstrating that deacetylation of H3K27ac occurs largely independent of Ubinucleins (Fig 7B). CUT&RUN analysis of H3K4me3 showed clear peaks over the TSS of active genes (Fig 7C). We observed a modest reduction after Xist induction in both WT and ΔUbn1/ΔUbn2 ESCs. Taken together our data show that removal of active marks over X-linked gene promoters is accomplished by Xist via Ubinuclein independent mechanisms.
We further performed directed ChIP for the X-linked genes Mecp2, Jarid1c, and Klf8 with antibodies specific for H3K27ac, H3K4me3, H3K27me3, and H2AK119ub. A small reduction of H3K4me3 and H3K27ac as well as an increase in H3K27me3 and H2Aub was evident in control ESCs after 48 hours of Xist induction (S6E Fig). In ΔUbn1/ΔUbn2 ESCs the reduction of H3K4me3 after Xist induction was variable. We also observed a markedly reduced increase in H3K27me3 after Xist induction compared to control ESCs. Taken together our findings demonstrate that loss of Ubinucleins causes a defect in establishment of H3K27me3 but deacetylation of H3K27ac over X-linked genes by Xist is unaffected.
Initial repression by Xist has no effect on the nucleosome free region at the TSS of X-linked genes
Our observation of a bimodal distribution of H3K27me3 around the TSS of active genes indicated the presence of a nucleosome free region [9]. Notably, this gap became more pronounced at X-linked genes when H3K27me3 increased after Xist induction. To investigate chromatin accessibility directly, we performed Assay for Transposase-Accessible Chromatin using sequencing (ATACseq) in WT and ΔUbn1/ΔUbn2 ESCs without and after 48 hours of Xist induction (Fig 7D). Induction of Xist slightly reduced overall accessibility over the X chromosome but not over autosomes in WT ESCs (S7A, S7B Fig) consistent with previous observations [39]. When the fold change in accessibility after Xist induction was plotted, a marginally smaller reduction was observed in ΔUbn1/ΔUbn2 compared to WT ESCs (S7A Fig, lower panel). ATAC-Seq coverage over X-linked EZH2 stations, CpG islands, CTCF sites, and regulatory regions showed only minimal changes of accessibility after Xist induction in WT and ΔUbn1/ΔUbn2 ESCs (S7C–S7E Fig).
ATAC-Seq profiles showed that the accessibility was similar over autosomes, but reduced over the X chromosome in ΔUbn1/ΔUbn2 compared to WT ESCs before Xist induction. The reduced read coverage is likely explained by the loss of the second X chromosome in ΔUbn1/ΔUbn2 ESCs (S3D Fig). Xist induction led to a moderate reduction of accessibility over the promoters and gene bodies of low and moderately expressed X-linked genes in WT and to a lesser extent in ΔUbn1/ΔUbn2 ESCs (S7F Fig). Autosomal genes were unaffected by Xist induction as expected (S7G Fig). Notably, a peak of high accessibility directly at the TSS persisted after Xist induction at a time when gene repression became evident in our RNA-Seq analysis (Figs 7D, and S7F). Therefore, a nucleosome free region at the promoter of X-linked genes is maintained during the initiation of XCI. We conclude that impairment of Xist function in ΔUbn1/ΔUbn2 ESCs is not explained by a requirement of HIRA complex mediated deposition of a nucleosome at the TSS of X-linked genes.
Discussion
Our study characterizes the function of the Ubinuclein genes in mouse development. The finding that Ubn1 is not essential in mice contrasts the earlier view of UBN1 as the principal Ubinuclein component of the HIRA complex [4]. We observe that homozygous Ubn1 mutant mice are healthy and fertile. Our mutation deletes a large N-terminal region of UBN1 protein that has been shown to be required for interactions with HIRA and histone H3.3 [10,17,32]. Although the remaining exons encoding the Ubinuclein middle domain, which mediates dimerization, are present, Western analysis does not detect a predicted truncated protein in liver nuclei of Ubn1 homozygous mice. In addition, combined mutations of Ubn1 and Ubn2 cause early embryonic lethality at a similar developmental stage as the Hira mutation. Taken together these observations strongly suggest that we have analyzed a loss-of-function mutation of Ubn1. The discrepancy to an earlier report finding Ubn1 essential for development of mouse zygotes [33] can likely be reconciled by considering differences in genetic background or methods for gene inactivation. Furthermore, acute loss of Ubn1 might have a more dramatic effect that might not resemble the constitutive mutation, and compensatory upregulation of Ubn2 in Ubn1 mutant embryos might be considered. However, we did not find compensatory regulation in Ubn1 mutant ESCs. Indeed, none of the mutations in HIRA complex components that we have analyzed affected expression of the remaining Hira, Ubn1, or Ubn2 genes. Our data support the view that, although Ubn1 and Ubn2 both contribute to mouse development, Ubn2 alone can support all functions of the HIRA complex in mice.
Phenotypic variation of the Ubn2 mutation causes embryonic lethality dependent on sex
We observe striking variations of the phenotypes of Ubn2 mutant embryos causing embryonic lethality with incomplete penetrance. Notably, the Ubn2 mutation causes a dramatic deficit of females in C57BL/6 mice. This is likely due to a fortuitous genetic situation allowing males, albeit at a lower than expected rate, to survive. Xist and Smchd1 have been the only genes associated with dosage compensation, whose mutation results in a female specific lethality [40,41]. Notably, preferential survival of males has also been observed due to anti-inflammatory activity of sex hormones in a study of mutations that impair DNA replication and lead to DNA damage [42]. McNairn et al. observe the first effect on sex ratios at E12.5. In contrast, a reduction of the number of female Ubn1^-/-^ Ubn2^-/-^ double mutant embryos occurs before E10.5 with embryos arresting before turning and, importantly, before sex hormones are produced. Similarly, the two phenotypic classes of the Hira mutation also arise before E10.5 [14]. We show that the severe class of the Hira mutant phenotype correlates with female sex, whereas the large majority of males associate with the mild class. Taken together with our observation on impaired Xist function in Ubn2, and Hira mutant ESCs our findings strongly suggest that female specific effects of mutations in HIRA complex genes are not the result of secondary sexual characteristics but of defects in XCI.
Mechanism of HIRA complex function in XCI
XCI is a multi-step process. Using inducible Xist expression in ESCs, we find that Hira and Ubinucleins are required for X-linked gene repression. Analysis of chromatin changes pinpoints which step in XCI requires HIRA complex function. Our data show that a repressive compartment is formed and Xist causes deacetylation of H3K27ac over promoters of X-linked genes in the absence of Ubinucleins. However, recruitment of H3K27me3 and H2AK119ub1 is impaired suggesting effects of loss of Ubinuclein genes on PRC1 and PRC2, respectively. A function of the HIRA complex in PRC2 recruitment has previously been observed at bivalent and developmentally regulated genes [16,43]. Although, gene silencing in XCI can occur without PRC2 function and in the absence of H3K27me3 [44,45], our observations indicate a defect in switching active X-linked gene promoters into a repressed configuration. Deacetylation of H3K27ac is likely mediated by HDACs independent of HIRA and Ubinucleins. One candidate is HDAC3 that is activated by SPEN following its recruitment by Xist repeat A [20,30,31]. We also find that a repressive compartment is formed in fibroblasts of female Ubn2^-/-^ embryos. Considering that the repressive compartment is formed over genomic repeats, which are not targeted by the HIRA complex, this observation is consistent with the role of Ubn1 and Ubn2 for histone H3.3 deposition in genes and regulatory elements [10].
Our study further defines the role of the HIRA complex in gene repression by Xist. We observe a consistent peak of accessibility in our ATACseq and H3K27me3 ChIP analysis before and after Xist induction independent of mutations in Ubinuclein genes. Therefore, occupation of the nucleosome free region is not an early step in the mechanism of gene repression of Xist that requires HIRA complex function. This is consistent with our previous analysis of the repression of Xist by the antisense Tsix RNA [46]. The nucleosome free region at the Xist promoter becomes occupied only when Tsix mediated repression becomes irreversible. However, we do not rule out an effect on dynamic positioning of nucleosomes around the TSS. Notably, in ESCs a well positioned -1 nucleosome is enriched in histone H3.3 and shows fast dynamics [9], and histone H3.3 has been suggested as a mark for the nucleosome free region at the TSS [47]. Further studies of histone H3.3 dynamics will be required to address this aspect fully. Considering that histone H3.3 is not enriched over silent genes [48], the HIRA complex is likely involved in switching an active to a repressed chromatin configuration at X-linked genes. Dynamic turnover of histones might be expected to contribute to the removal of active chromatin modification. However, we find that deacetylation of H3K27ac is independent of HIRA complex function and removal of active marks is unaffected in ΔUbn1/ΔUbn2 ESCs. An interesting possibility is that gene repression by Xist might depend on a specific feature of histone H3.3. Histone H3.3 but not H3.1 and H3.2 possesses a serine at position 31, which can be phosphorylated and has been implicated in gene regulation [49,50]. Therefore, it is enticing to speculate that serine 31 might play a role in the mechanism of gene repression by Xist.
A striking observation of our study is the partial loss of Xist function in the absence of Hira or Ubinucleins. This finding indicates the existence of parallel pathways at the initiation of XCI. One potential candidate is HDAC3. A recent study has observed that HDAC3 is already present on the X chromosome before Xist induction and a partial loss of gene repression is observed in ESCs with an Hdac3 mutation [31]. Our observation that deacetylation of H3K27ac at X-linked gene promoters does not require Ubinucleins is consistent with a parallel pathway supporting the residual silencing function of Xist. Conceivably, HIRA complex function might also be partially compensated by other nucleosome assembly complexes. Histone H3.3 deposition is mediated by ATRX and DAXX at the centromeres and telomeres [51]. Notably, ATRX associates with the Xi in somatic and trophoblast stem cells [52]. However, Atrx has been implicated in the maintenance of XCI and not the initial establishment of gene repression by Xist. Our mutant ESC and mouse lines will facilitate investigation of potential redundancy between Hira or Ubinucleins, and Atrx or Daxx in future studies.
Materials and methods
Ethics statement
All animal experiments were approved by the Cantonal Veterinary office, Waltersbachstrasse 5, 8090 Zurich, Switzerland and carried out under the cantonal permit number ZH153/2021 and national permit number 34039 in accordance with Swiss federal and cantonal regulations.
Culture and gene editing of ESCs
HATX3 and HATX8 ESCs were cultured as described previously [20]. Xist was induced by adding 1 μg/ml doxycycline. gRNAs were designed using the CHOPCHOP [53], GuideScan [54], and E-CRISPR [55] and cloned into pX458 (Addgene, #48138). 2µg of the vector was transfected into 300000 ESCs using Lipofectamin 2000 (ThermoFisher). GFP positive ESCs were sorted into 96-wells and genotyped by PCR and Sanger sequencing. An HA tag was inserted into Ubn2, using 1µg of ssDNA donor template (5’-tttcttttctcagatggaggccaaagtaaaggggacactaagttaccacggaaacctcagTCCGCCTGGTCCCACCCTCAGTTTGAGAAAGGCGGCGGCTACCCCTACGACGTGCCCGACTACGCCTGActttccagcaagggggagaggaaccacttggctggctggcgggaccgacctgatgggaag-3’). The Hira coding region was amplified from cDNA and cloned into a PiggyBac (Pb) vector containing an EF1-alpha promoter and a Ruby fluorescence reporter (System Biosciences, Cat No. PB531A-2). The Ubn2 cDNA (Dharmacon, #MMM1013-202770327) was cloned into the PiggyBac vector described above. The ΔBLLF1 deletion was introduced by cutting the Ubn2 expression vector with two Cas9 RNPs.
Gene editing in mice
20 pmol Cas9 protein mixed with 40 pmol of sgRNA in injection buffer (8 mM TrisHCl pH 7.4, 0.1 mM EDTA) adjusted to a final volume of 12.5 µl was used for pronuclear microinjection of C57BL/6 zygotes. Mutations were identified by PCR using DNA from ear biopsies from founder mice.
RNA isolation and RNA-Seq analysis
Total RNA was isolated using RNeasy Mini Kit (Quiagen, #74104) and DNA removal by on-column DNAse I digestion (Qiagen, #79254). 500 ng total RNA were used for cDNA synthesis using the PrimeScript RT Master Mix kit (Takara, #RR036A). Real-time quantitative PCR was performed using SYBER Green on a LightCycler 480 system (Roche). RNA-Seq was performed in triplicates for Xist inducible WT, ΔUbn1, ΔUbn2, ΔUbn1/Ubn2, and ΔHira ESCs non-treated and treated with Dox for 48h. Total RNA was isolated as above and used for NGS library preparation with the Illumina TruSeq Stranded mRNA kit. Sequencing was performed on a Novaseq 6000 (Illumina, Inc, California, USA) using single-end 100 bp sequencing.
Co-Immunoprecipitation
2 mg of ESC lysate was incubated over night at 4^o^C on a rotator with 250U of Benzonase nuclease (Sigma-Aldrich, #E1014) and 55 µl (dry volume) of pre-washed anti-HA affinity matrix (Roche, #11815016001) in a final volume of 1 ml RIPA buffer. Subsequently, beads were washed twice for 5 min under rotation with wash buffer (50 mM TrisHCl pH 8, 150 mM NaCl, 1 mM EDTA, 0.02% NP40), and once with PBS. Proteins were eluted from the beads by resuspending in 2 x Laemmli buffer and boiling for 5 min at 95^o^C.
Immunofluorescence and RNA FISH analysis
For immunofluorescence staining, cells were grown on multiwell Roboz Slides (Cellpoint Scientific, USA), and then fixed and permeabilized. Primary antibodies were diluted in blocking buffer and incubated over night at 4^o^C followed by incubation with secondary antibodies for 1h at RT. Nuclei were counterstained with 1.4 μM DAPI (Molecular Probes, #D1306). After several washes in PBS/ 0.1% Tween-20 (PBST) slides were mounted using Vectashield (Reactolab, #H1000), or Mowiol (Sigma-Aldrich, #81381). For combined immunofluorescence RNA FISH analysis, incubation with primary and secondary antibody was 45 min at room temperature using RiboLock RNAse inhibitor (Thermo Scientific, #EO0381). Slides were postfixed, air dried, and hybridized with Cy3-labeled Xist RNA FISH probe (Cy3 dCTP, GE Healthcare Amersham) generated by random priming with Prime-it II, (Stratagen, #300385) and competed with mouse Cot-1 DNA (Invitrogen, #18440-016), salmon sperm DNA (Invitrogen, #15632-011) and tRNA (Invitrogen, #AM7119) in Hybrisol VII (MP Biomedicals). After overnight incubation, and washing, nuclei were counterstained with a 1.4 μM DAPI solution (Molecular Probes, #D1306).
Native ChIP and ChIP-Seq analysis
All buffers contained Complete Mini protease inhibitor (Roche, #11836153001) and 5 mM Na-butyrate deacetylase inhibitor. 12 million ESCs were pelleted and resuspended in 90 μl of Lysis Buffer (LB) (50 mM TrisHCl pH 7.5, 150 mM NaCl, 0.1% sodium deoxycholate, 1% Triton, 5 mM CaCl2). Chromatin was digested using 60 μl of LB containing 0.3 μl Micrococcal nuclease (Cell signalling, #10011S) at 37^o^C for 15 min. Reactions were stopped with 15 μl 0.5 M EDTA, and Stop Buffer (SB) (50 mM TrisHCl pH 7.5, 150 mM NaCl, 0.1% sodium deoxycholate, 1% Triton, 30 mM EGTA, 30 mM EDTA). After adjusting the volume to 1 ml with LB/ SB (1:1), 30 μl were used as input. 300 μl of chromatin was adjusted to 1ml with LB/ SB (1:1) and incubated over night with corresponding antibody at 4^o^C. 15 μl Dynabeads protein G (ThermoFisher, #10004D) blocked with blocking buffer (PBS 1X, 0.5% Tween, 0.5% BSA) were added and incubated 2h at 4^o^C. After washing, DNA was eluted from beads and purified using the MinElute PCR purification kit (Qiagen, #28004). ChIP-Seq was performed in triplicates for Xist inducible WT and ΔUbn1/Ubn2 ESCs, witout or after 48h of Xist induction. Chromatin was prepared as above with digestion with Micrococcal nuclease for 20 min. 1 ng DNA was used for library preparation using the NEBNext ultra II DNA Library Prep Kit from NEB (New England Biolabs (NEB), Ipswich, MA, #7103). Libraries were size-selected for fragments of 300–600 bp with Agencourt AMPure XP magnetic beads (Beckman Coulter), and sequenced on an Illumina HiSeq 2500 machine using 125 bp single-end sequencing.
CUT&RUN analysis
CUT&RUN was performed using CUT&RUN Assay Kit (Cell SignalingTechnology #86652) and antibodies against H3K27ac (Cell Signaling Technology #8173, 1:50) and H3K4me3 (Cell Signaling Technology rabbit mAb #9751, 1:50). DNA was purified from enriched chromatin samples using Spin Columns (Cell Signaling Technology #14209). For library prep, NEBNext Ultra II DNA Library Prep Kit for Illumina (NEB #E7103) was used. Libraries were sequenced on an Illumina MiSeq or Illumina NextSeq 2000 instrument.
ATACseq
ATACseq was performed in triplicates following the improved version of the original protocol [56]. ESCs were lysed, and nuclei incubated with TDE1 tagment DNA enzyme (Illumina, #15027865) in tagment DNA buffer (Illumina, #15027866). DNA was purified with MinElute PCR purification kit, amplified and multiplexed with Nextera DNA CD indexes (Illumina, #20015881). Libraries were purified with MinElute PCR purification kit and used for paired-end 75 bp sequencing on a HS 2500 illumina instrument.
Computational analysis of next generation sequencing data
For the RNAseq analysis, Trimmomatic [57] and HISAT2 [58] were used for pre-processing of the reads including trimming of adaptors and alignment of reads to the mm10 mouse genome reference assembly, respectively. HTSeq was used for counting RNAseq reads that overlap gene features, handling of genome annotation, and performing data analysis involving genomic coordinates [59]. Data was normalized to transcripts per million (TPM) for all analyses except for differential gene expression. DESeq2version 1.38.3 was used for the analysis of differential gene expression and calculation of adjusted p-values from raw read counts of RNAseq datasets [35]. For the analysis of ChIPseq, ATACseq, and CUT and RUN datasets Trimmomatic [57] and Bowtie2 [60] were used for pre-processing of the reads including trimming of adaptors and alignment of reads to the mm10 mouse genome reference assembly, respectively. HTSeq was used for counting reads that overlap gene features, handling of genome annotation, and performing data analysis involving genomic coordinates [59]. Python and R scripts, genomic annotation, and intermediate data for computational analyses are made available on github at https://github.com/WutzLab.
Statistics
Paired, parametric, two-tailed t-tests were performed to determine significance with a p-value threshold smaller than 0.05. Calculations were performed using Prizm Graphpad Version 7.
Supporting information
S1 TextIncludes the Supplementary methods, S1 Table legend, S2 Table legend, S4Table legend, and Supplementary References.(PDF)
S1 FigContains S1 Fig and legend.(PDF)
S2 FigContains S2 Fig and legend.(PDF)
S3 FigContains S3 Fig and legend.(PDF)
S4 FigContains S4 Fig and legend.(PDF)
S5 FigContains S5 Fig and legend.(PDF)
S6 FigContains S6 Fig and legend.(PDF)
S7 FigContains S7 Fig and legend.(PDF)
S1 TableIncludes S1 Table showing numbers and statistics of Ubn2 mutant mouse crosses.(XLSX)
S2 TableIncludes S2 Table showing numbers and statistics of Ubn1 and Ubn2 double mutant mouse crosses.(XLSX)
S3 TableIncludes S3 Table showing SILAC mass spectrometry data of HA-Ubn2 interactors.(PDF)
S4 TableIncludes S4 Table as an archive of 4 Excel workbooks with lists of differentially regulated genes in ΔUbn1, ΔUbn2, ΔUbn1/ΔUbn2, and ΔHira mutant compared to WT ESCs.(ZIP)
S5 TableIncludes S5 Table showing sequences of primers and gRNAs used in this study.(PDF)
S6 TableIncludes S6 Table showing a list of reagents, instruments, and materials used in this study.(PDF)
S1 DataContains source data for graphs in Fig 4.(XLSX)
S2 DataContains source data for Western blots in Fig 4.(PDF)
S3 DataContains source data for graphs in Fig 5.(XLSX)
S4 DataContains source data for graphs in Fig 6.(XLSX)
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1TornéJ, Ray-Gallet D, Boyarchuk E, Garnier M, Le Baccon P, Coulon A, et al. Two HIRA-dependent pathways mediate H 3.3 de novo deposition and recycling during transcription. Nat Struct Mol Biol. 2020;27(11):1057–68. doi: 10.1038/s 41594-020-0492-7 32895554 · doi ↗ · pubmed ↗
- 2Bouvier D, Ferrand J, Chevallier O, Paulsen MT, Ljungman M, Polo SE. Dissecting regulatory pathways for transcription recovery following DNA damage reveals a non-canonical function of the histone chaperone HIRA. Nat Commun. 2021;12(1):3835. doi: 10.1038/s 41467-021-24153-1 34158510 PMC 8219801 · doi ↗ · pubmed ↗
- 3Hinchcliffe EH, Day CA, Karanjeet KB, Fadness S, Langfald A, Vaughan KT, et al. Chromosome missegregation during anaphase triggers p 53 cell cycle arrest through histone H 3.3 Ser 31 phosphorylation. Nat Cell Biol. 2016;18(6):668–75. doi: 10.1038/ncb 3348 .27136267 · doi ↗ · pubmed ↗
- 4Delaney K, Weiss N, Almouzni G. The cell-cycle choreography of H 3 variants shapes the genome. Mol Cell. 2023;83(21):3773–86. doi: 10.1016/j.molcel.2023.08.030 37734377 PMC 10621666 · doi ↗ · pubmed ↗
- 5Choi J, Kim T, Cho E-J. HIRA vs. DAXX: the two axes shaping the histone H 3.3 landscape. Exp Mol Med. 2024;56(2):251–63. doi: 10.1038/s 12276-023-01145-3 38297159 PMC 10907377 · doi ↗ · pubmed ↗
- 6Goldberg AD, Banaszynski LA, Noh K-M, Lewis PW, Elsaesser SJ, Stadler S, et al. Distinct factors control histone variant H 3.3 localization at specific genomic regions. Cell. 2010;140(5):678–91. doi: 10.1016/j.cell.2010.01.003 20211137 PMC 2885838 · doi ↗ · pubmed ↗
- 7Pchelintsev NA, Mc Bryan T, Rai TS, van Tuyn J, Ray-Gallet D, Almouzni G, et al. Placing the HIRA histone chaperone complex in the chromatin landscape. Cell Rep. 2013;3(4):1012–9. doi: 10.1016/j.celrep.2013.03.026 23602572 PMC 3974909 · doi ↗ · pubmed ↗
- 8Garrick D, Sharpe JA, Arkell R, Dobbie L, Smith AJH, Wood WG, et al. Loss of Atrx affects trophoblast development and the pattern of X-inactivation in extraembryonic tissues. P Lo S Genet. 2006;2(4):e 58. doi: 10.1371/journal.pgen.0020058 16628246 PMC 1440874 · doi ↗ · pubmed ↗