Identification of pathogens causing fever in returning travelers using next-generation sequencing
Kenji Gotoh, Nobuyuki Hamada, Takahito Kashiwagi, Koyu Hara, Hiroshi Watanabe

TL;DR
This study used next-generation sequencing to identify pathogens causing fever in returning travelers when traditional methods failed, finding that common coronaviruses like HCoV-OC43 were often responsible.
Contribution
The study demonstrates the utility of NGS in diagnosing undiagnosed febrile illnesses in returning travelers, highlighting the role of common coronaviruses.
Findings
NGS detected HCoV-OC43 in all eight samples from undiagnosed febrile returning travelers.
Outpatient follow-up is viable for febrile travelers if tropical diseases are excluded and their condition is stable.
Conventional RT-PCR confirmed HCoV-OC43 in two of the eight NGS-positive samples.
Abstract
In cases of fever following international travel, medical evaluation should consider the prevalence of infectious diseases in the travel destination. However, there are instances where a definitive diagnosis cannot be made. Identifying these unknown pathogens is crucial for managing febrile returning travelers and as a model for the early detection of emerging infectious diseases. The aim of this study was identification of pathogens from febrile cases where fever was the primary symptom and no other specific clinical features were present. Between 2008 and 2020, a total of 164 travelers visited Kurume University Hospital due to illness after returning from abroad. However, despite extensive testing, no definitive diagnosis was reached for 18 febrile travelers. Next-generation sequencing (NGS) was performed on eight samples (five whole blood, one serum, one cerebrospinal fluid, and one…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
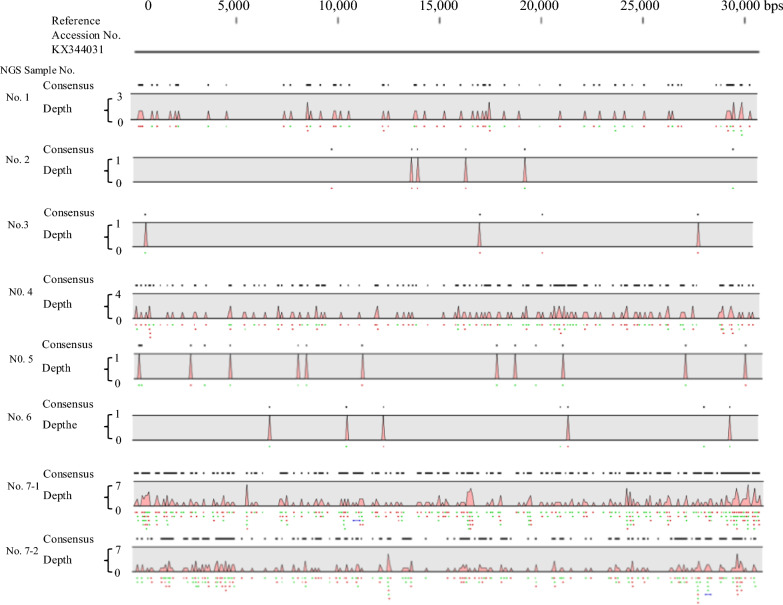 Figure 1
Figure 1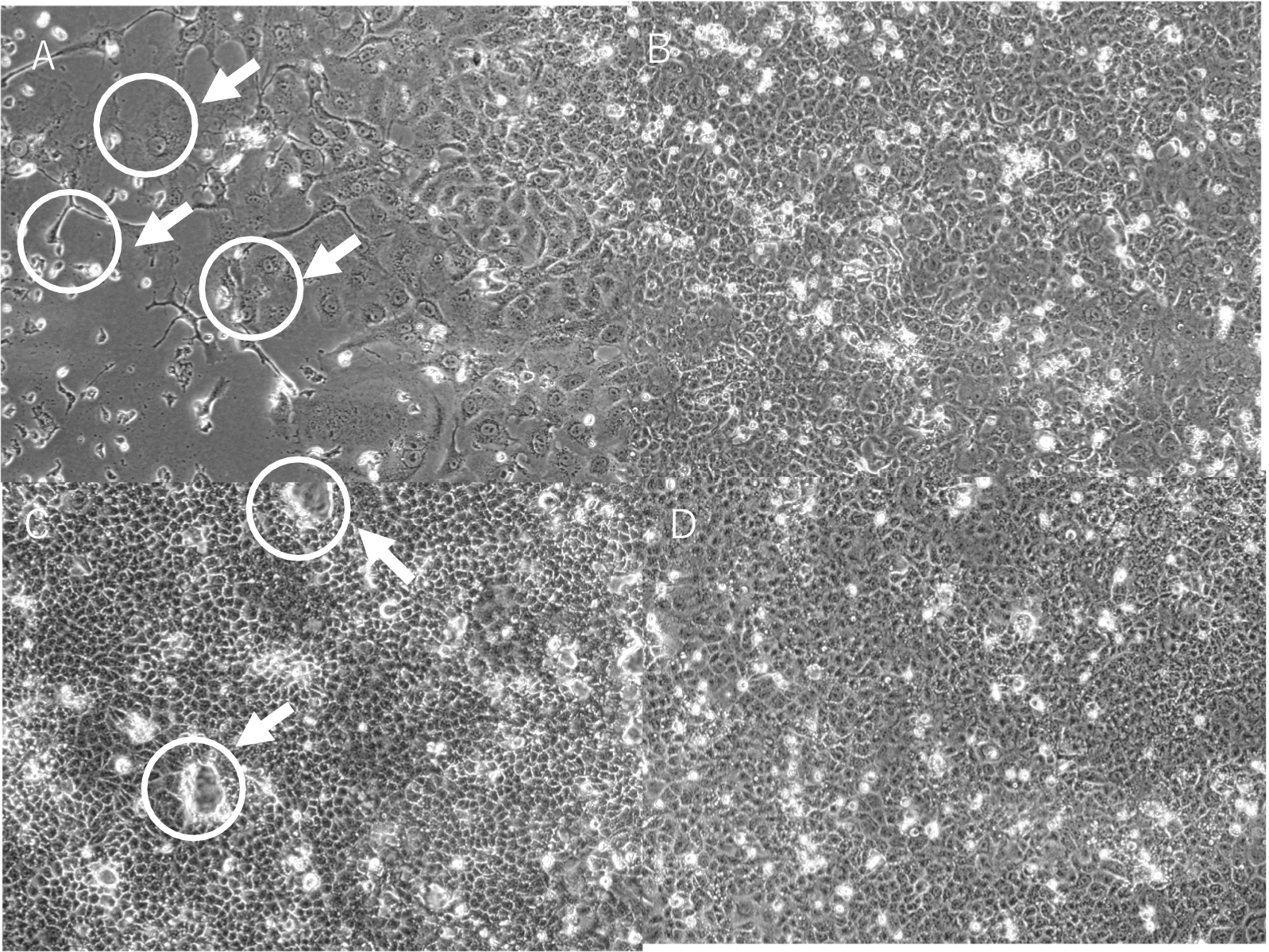 Figure 2
Figure 2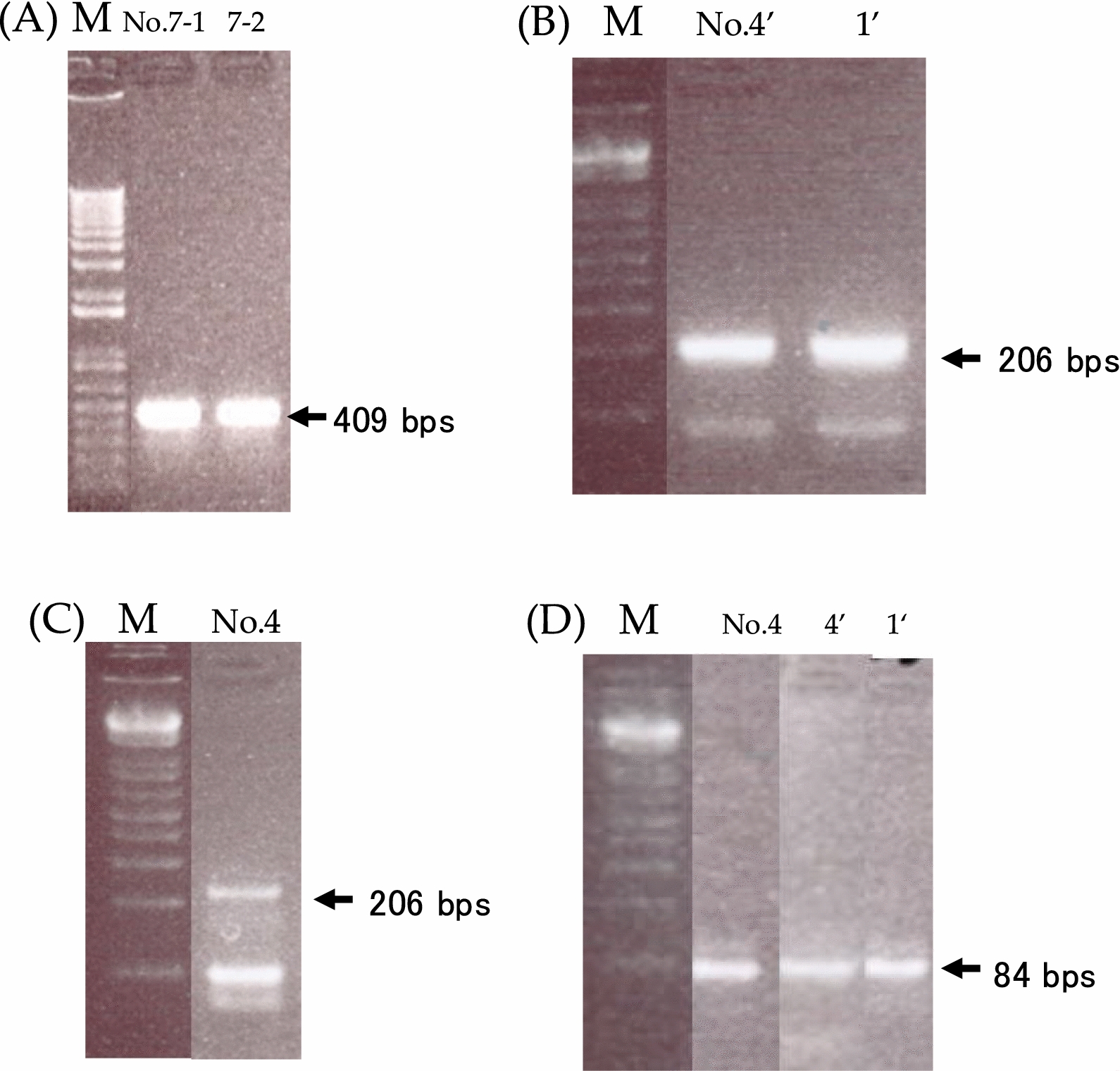 Figure 3
Figure 3Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsTravel-related health issues · Malaria Research and Control · Mosquito-borne diseases and control
Background
In recent years, the number of international travelers has increased significantly, leading to a rise in the number of travelers developing illnesses [1, 2]. Furthermore, the distribution of vector-borne diseases, including Japanese encephalitis, has been expanding, likely due to global environmental changes [3, 4]. Another serious concern is the spread of antimicrobial-resistant bacteria and infectious diseases by foreign travelers [5, 6]. Travel to tropical regions and developing countries, in particular, carries a high risk of infectious diseases, and many returning travelers present with fever of unknown origin [7, 8].
Before the COVID-19 pandemic, there was a significant increase in both the number of Japanese travelers going abroad and foreign travelers visiting Japan, which in turn increased the risk of travelers contracting various infectious diseases [9].
Recently, next-generation sequencing (NGS) has been used to analyze febrile returning travelers, leading to the detection of various viruses, such as dengue virus, Epstein–Barr virus, hepatitis A virus, hepatitis B virus, hepatitis C virus, hepatitis E virus, and chikungunya virus [10, 11]. However, the types and the distribution of infectious diseases vary by region [12], and detailed analyses of where and which infections travelers contract are still insufficient. Additionally, many cases of post-travel fever remain undiagnosed. Analyzing these cases in detail could contribute to better infection control guidance and recommendations for vaccinations during pre-travel medical counseling. Furthermore, accumulating such research could aid in the early detection of emerging infectious disease pathogens like COVID-19 and in predicting pathogens that could cause the next pandemic.
In this study, we report our attempt to identify the causative microorganisms through NGS analysis of samples from patients who developed fever after traveling abroad but could not be diagnosed despite extensive pathogen testing.
Materials and methods
Ethical approval
All studies described herein were approved by the Human Ethics Review Board of Kurume University (Approval No. 20263).
Analysis of returning travelers
We retrospectively reviewed the medical records of returning travelers who visited Kurume University Hospital between 2008 and 2020 and classified them based on factors, such as age, sex, nationality, travel history, symptoms, and clinical diagnosis. We extracted cases of febrile patients whose cause of fever could not be identified despite routine medical diagnostic tests, including blood tests, serological tests, antigen tests, and nucleic acid amplification tests. Whole blood, serum, and throat swab samples were collected from all patients, and additional specimens were obtained as needed based on the patients'symptoms. Stool samples were collected from patients presenting with gastrointestinal symptoms such as diarrhea or abdominal pain. Depending on the travel destination and clinical presentation, a selection of relevant diagnostic tests is performed. Table 1 lists all the diagnostic tests that were conducted at our institution.Table 1. Laboratory tests performed on cases of fever after traveling abroadRapid test Malaria Antigen, Malaria Ag. pLDH/HRP2, Premier Medical Corporation, USA Dengue virus antigen, OnSite Dengue Ag Rapid Test, CTK Biotech Inc, USA Dengue virus antibody, OnSite Dengue Ag Rapid Test, CTK Biotech Inc, USA Chikungunya virus antigen, OnSite Dengue Ag Rapid Test, CTK Biotech Inc, USA Chikungunya virus antibody, OnSite Dengue Ag Rapid Test, CTK Biotech Inc, USAPCR test Adeno virus Chikungunya virus Dengue virus Enterovirus Hepatitis E virus Malaria Rhinovirus West nile virus Zika virus
NGS analysis
Samples that were not appropriately managed or had insufficient volume for NGS analysis were excluded. DNase treatment was not performed; therefore, the extracted samples contained not only RNA but also DNA. Additionally, since DNA viruses involved in active infections are generally replicating and producing RNA transcripts, we considered RNA samples appropriate for pathogen detection. Total RNA was extracted from eight unidentified clinical samples using the QIAamp Viral RNA Mini Kit (Qiagen, Hilden, Germany). Double-stranded cDNA was synthesized using the PrimeScript Double-Strand cDNA Synthesis Kit (TAKARA, Tokyo, Japan). A NGS library was prepared using the Nextera XT Library Prep Kit (Illumina, San Diego, CA, USA) according to the manufacturer’s protocol. NGS was performed using the Illumina MiSeq platform. Host reads were removed by mapping the raw reads to the human reference genome GRCh38 (Genome Reference Consortium Human Build 38). Reads that mapped to the human genome were discarded prior to pathogen detection analysis. The sequencing results were then mapped against reference sequences containing various viral genomes using the CLC Genomics Workbench (Qiagen, Hilden, Germany).
Virus isolation and RNA extraction
Virus isolation was performed on samples from cases in which the cause of fever had not been identified and sufficient sample volume was available (No. 1 and No. 4). The samples were diluted tenfold and inoculated onto VeroE6 cell monolayers, then cultured in Eagle’s minimum essential medium (MEM) supplemented with 2% fetal bovine serum (FBS) and kanamycin (final concentration: 50 µg/mL) for one week. A single blind passage was performed. Cells exhibiting cytopathic effects (CPE) were frozen and thawed as needed. Total RNA was extracted from cell suspensions using TRIzol™ reagent (Thermo Fisher Scientific, Waltham, MA, USA) according to the manufacturer’s protocol and stored at − 80 °C.
RT-PCR for confirmation
To reconfirm the NGS results and identify viruses in CPE-positive samples (No. 1 and 4), conventional RT-PCR was performed. RNA samples were re-purified using TRIzol™ reagent, and PrimeScript™ RTase (Takara, Japan) and Takara EX Taq (Takara, Japan) were used for reverse transcription and PCR, respectively. PCR was conducted under identical cycling conditions with different primers (Table 2). The cycle conditions were as follows: 45 ℃ for 1 h (RT step) followed by 94 ℃ for 2 min; 25 cycles at 94 ℃ for 20 s, 60 ℃ for 20 s, and 72 ℃ for 20 s (PCR step). RNA samples were stored at − 80 °C. RT-PCR was also performed for sample No. 7–2, for which residual RNA was available. RNA samples corresponding to NGS numbers 2, 3, 5, and 6 were unavailable and therefore not analyzed.Table 2. Primers for RT-PCR of Human coronavirus OC43NoAmplicon size (bps)Name of primerSequence14095KYOC43FGATGAGTCCTTTTACAAGAACATG25KYOC43RTAATCATCCACATCGGTCCATTTAC3206coronapolOUTFTTTGAGTGATGATGGGGTTGTGTGTTATAATTCTG4coronapolOUTRCACCATCCATCTTTACAAGCATTGTGTGTTGTGAA584coronapolINFGTGCCTTTCAACAGGTATTATATTATCAAAATAAC6coronapolINRCATTATTTATGTCATGTTCAACCCAACATTTGGAT^^GTGCCTTTCAACAGGTATTATATTATCAAAATAACGTTTTTATGTCAGAATCCAAATGTTGGGTTGAACATGACATAAATAATG (NGS No. 4, No. 4–2/Vero, and NGS No. 1–2/Vero)
Results
Characteristics of returning travelers
A total of 164 returning travelers (149 Japanese and 15 foreigners) visited Kurume University Hospital during the study period. Their mean age was 35.7 years (range: 11 months to 79 years), and 79 were male. Diagnoses and travel destinations are shown in Table 3. The most common diseases included 37 cases of animal bites with rabies risk, 33 cases of traveler’s diarrhea, 21 cases of upper respiratory tract infections, 12 cases of dengue fever, and 12 cases of influenza A or B. Of the 12 dengue cases, 11 were from travelers returning from Southeast Asia, and 5 of the 7 malaria cases were diagnosed in travelers returning from Africa. However, 18 febrile returning travelers remained undiagnosed despite undergoing a comprehensive set of diagnostic tests, including rapid tests and PCR for tropical and viral infections, as summarized in Table 1. These cases highlight the diagnostic limitations of conventional testing approaches.Table 3. The diagnoses and destinations of returning travelersDiagnosisNo. of patientsSoutheast AsiaSouth AsiaEast AsiaMiddle EastEuropeAfricaNorthernAmericaCentral and South AmericaOceaniaAnimal bite with rabies risk372444120020Traveler's diarrhea331693103100Upper respiratory tract infection211250001111Dengue fever121110000000Influenza A or B12801002010Malaria7020005000Lower respiratory tract infection7301012000Amoebiasis3300000000Cellulitis2100000010Giardiasis2110000000Taenia saginata2100010000Lyme disease2000020000Aseptic meningitis2100000010Typhoid fever1010000000Biliary tract infection1000000010Cutaneous leishmaniasis1000001000Rash (unknown origin)1010000000Fever (unknown origins)181301013000Total1649424102717271
Characteristics of viral analysis cases
Of the 18 undiagnosed cases, 8 samples from 7 cases were analyzed as they had been appropriately stored and had sufficient specimen volume for NGS. These travelers (3 males and 4 females, aged 5–49 years) had returned from Southeast Asia, South Asia, and Central America. Whole blood, serum, cerebrospinal fluid, or nasopharyngeal swabs were used for viral analysis.
NGS analysis
NGS detected human coronavirus OC43 (HCoV-OC43) genes in 6 of the 8 samples, HCoV-OC43 and herpes simplex virus type 1 (HSV-1) in 1 sample, and HCoV-OC43 and mumps virus in another sample (Table 4). As shown in Fig. 1, the NGS sequences of patients were identical to the reference genome of the human coronavirus OC43 strain (accession number: KX344031).Table 4. Characteristics of febrile returning travelers and virus analysisNGS* NoAgeCollection Date (year/month/date)SexTravel historySymptomSample typeDetected gene by NGSConfirmed gene by RT-PCRVirus isolation14923-03-2016MaleIndiafeverserumHCoV-OC43 and Herpes simplex virus-1**HCoV-OC43 (via virus isolation)HCoV-OC432807–08-2013FemaleIndonesiafeverwhole bloodHCoV-OC43not donenot done31905–09-2012FemaleThailandfeverwhole bloodHCoV-OC43not donenot done42615-03-2016FemaleHaiti, Jamaicafever, headachewhole bloodHCoV-OC43HCoV-OC43 (direct & via virus isolation)HCoV-OC4354618-10-2011MaleVietnamfever, headachewhole bloodHCoV-OC43not donenot done64101–09-2011FemaleCambodiafeverwhole bloodHCoV-OC43not donenot done7–1530-11-2011MaleIndonesiafever, headachecerebrospinal fluidHCoV-OC43 and Mumps virusHCoV-OC43not done7–230-11-2011nasopharyngeal swabHCoV-OC43HCoV-OC43not done^^next-generation sequencing^**^CCGCCCGTCTCGTCCAGAAGACCCCGGCGTACCCAGCACCFig. 1Coverage of next-generation sequencing (NGS) reads from patient samples mapped to the reference genome of the human coronavirus OC43 strain (accession number: KX344031). The upper solid line represents the reference genome. The second, discontinuous line from the top indicates the consensus sequence. Sequence depth across the genome (ranging from 0 to 7 nucleotides) is represented by red (forward strand) and green (reverse strand) dots beneath the red waveform. The red waveform also reflects read depth but is partially omitted for clarity and scale. Depth shows number of reads
Virus isolation
Virus isolation was performed using serum from sample NGS* No.1 and whole blood from sample NGS* No.4. As shown in Fig. 2, cells inoculated with samples NGS No.1 and NGS No.4 exhibited syncytia formation after 7 and 5 days, respectively.Fig. 2. VeroE6 cells inoculated with second-passage NGS samples showing cytopathic effects (CPE). A: Cells inoculated with sample from NGS Case No. 1, seven days post-inoculation. B: Negative control corresponding to (A). (C): Cells inoculated with sample from NGS Case No. 4, five days post-inoculation. D: Negative control corresponding to (C). Syncytium formation, a typical CPE, was observed in (A) and (C)
RT-PCR for confirmation
Based on NGS and virus isolation results, RT-PCR was performed only on NGS*No.1 and No.4, for which virus isolation was successful, and on No.7, for which residual RNA was available (Table 4). PCR was conducted under identical cycling conditions with different primers. The cycle conditions were as follows: 45 ℃ for 1 h (RT step) followed by 94 ℃ for 2 min; 25 cycles at 94 ℃ for 20 s, 60 ℃ for 20 s, and 72 ℃ for 20 s (PCR step). The viruses detected via NGS in samples No.1, No.4 and No.7 were confirmed by RT-PCR (Fig. 3).Fig. 3.A 1% agarose gel electrophoresis of conventional RT-PCR products. The arrow indicates the expected size of the RT-PCR product (409 base pairs). Lane 1: NGS No. 7–1; Lane 2: NGS No. 7–2. The numeric suffix refers to the sample type (see Table 4). (B–D) 3% agarose gel electrophoresis (NuSieve™ 3:1) of conventional RT-PCR products. B Lane 1: NGS No. 4’ from isolated virus; Lane 2: NGS No. 1’ from isolated virus. No bands were observed using 1 µL of each sample. C Repeated PCR using the same samples with increased volume. Lane 1: NGS No. 4. The arrow indicates the RT-PCR product size (206 base pairs). D Lane 1: NGS No. 4; Lane 2: NGS No. 4 ‘from isolated virus; Lane 3: NGS No. 1’ from isolated virus. The arrow indicates the amplicon size (84 base pairs). “M” indicates the size marker
Discussion
As in previous reports, most post-travel medical visits involved cases of animal bites, traveler’s diarrhea, dengue fever, and influenza. In this study, we detected HCoV-OC43 in all tested samples collected from patients with fever of unknown origin. HCoV-OC43 is generally recognized as a cause of respiratory and gastrointestinal infections [13, 14]. Our findings suggest that in cases presenting only with prolonged fever after travel, HCoV-OC43, typically associated with the common cold, may be involved. If tropical diseases, such as malaria, dengue fever, and typhoid fever, can be excluded based on travel history, it may be reasonable to manage such cases as common colds.
Although this study was conducted before the COVID-19 pandemic, our findings suggest that various coronaviruses were already prevalent in Asia.
Our study was conducted at a single center in Kyushu, Japan, which is geographically close to Southeast Asia, and the number of cases was limited; therefore, our findings do not fully reflect the overall situation of febrile illnesses after travel in Japan. In addition, because multiple pathogen genomes were sometimes detected by NGS in a single case, it is not always possible to determine whether each detected pathogen was truly responsible for the fever. Continued epidemiological monitoring of febrile post-travel patients is necessary.
Conclusion
HCoV-OC43 was detected in all tested cases of prolonged fever in travelers returning to Japan, suggesting it may be a common cause of post-travel febrile illness. This finding highlights its clinical relevance and supports the continued use of NGS for improving diagnosis and management in travel clinics.