Coding-complete genome sequence of a divergent member of the genus Gordisvirus detected in Sabethes (Peytonulus) undosus Coquillet mosquitoes (Diptera: Culicidae) from Brazil
Daniel Dias, Bruna Nascimento, Ana Cruz, Sandro Silva, Lúcia Reis, Fábio Silva, Lucas Silva, Sâmia Silva, Durval Vieira, Roberto Brandão, José Junior, Alessandra Santos, Hanna Reis, Joaquim Neto

TL;DR
This paper presents the full genome sequence of a new virus found in mosquitoes from the Brazilian Amazon.
Contribution
The study provides the first complete genome sequence of a divergent Gordisvirus.
Findings
The virus was identified in Sabethes (Peytonulus) undosus mosquitoes.
The genome is 12,150 nucleotides long and contains five open-reading frames.
Phylogenetic analysis confirmed its classification within the genus Gordisvirus.
Abstract
We report the complete genome sequence of a divergent member of the genus Gordisvirus (family Xinmoviridae, order Mononegavirales), obtained through metagenomic sequencing of Sabethes (Peytonulus) undosus Coquillett mosquitoes in the Brazilian Amazon. Phylogenetic analyses confirmed its classification. The genome comprises 12,150 nucleotides and encodes five open-reading frames.
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
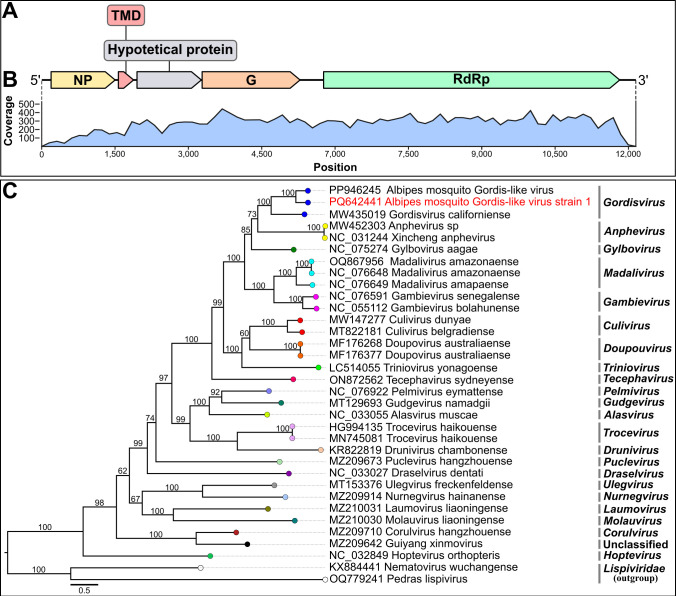 Fig 1
Fig 1Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsMosquito-borne diseases and control · Insect symbiosis and bacterial influences · Plant Virus Research Studies
ANNOUNCEMENT
Xinmoviridae is a family of negative-sense RNA viruses with genomes of 9 to 14 kilobases that typically infect arthropods (1). Virome studies in mosquitoes have expanded the knowledge about Xinmovirids, leading to the identification of several novel viruses (2, 3). Here, we report the coding-complete genome sequence of a divergent member of the genus Gordisvirus, recovered through metagenomic sequencing of a pool of ten female Sabethes (Peytonulus) undosus Coquillet collected in 2022 in Belém, Pará, Brazilian Amazon.
The pool containing whole insect bodies was homogenized in 500 µL of Dulbecco’s Phosphate Buffered Saline (Gibco, Massachusetts, USA), with 2% penicillin and streptomycin (Sigma-Aldrich, Massachusetts, USA; Cat.No. P4333), 1% Amphotericin B (Sigma-Aldrich; Cat.No. A2942), 5% fetal bovine serum (LGC Biotecnologia, São Paulo, Brasil; Cat.No.10-bio500), and a 3 mm tungsten bead (Qiagen, Hilden, Germany) using TissueLyser II (Qiagen) at 25 Hz for 1 min. Viral RNA was extracted using the QIAamp Viral RNA kit (Qiagen). cDNA synthesis was performed with the SuperScript (Agilent Technologies, California, USA) and NEBNext (New England Biolabs, USA) kits. Libraries were prepared with the SureSelectQXT kit (Agilent) and sequenced by the paired-end method on the NextSeq 550 platform (Illumina, California, USA) using the NextSeq 500/550 High Output kit v.2.5 (300 cycles).
Quality control of reads was performed using Fastp v.0.23.4 (4). De novo assembly was conducted using MEGAHIT v.1.2.9 (5). Contigs were compared to the RefSeq viral database (https://ftp.ncbi.nlm.nih.gov/refseq/release/viral/) using DIAMOND v.2.1.10 (6) and inspected with MEGAN v.25.10 (7). Open reading frames (ORFs) were predicted using NCBI ORFfinder (https://www.ncbi.nlm.nih.gov/orffinder/) and annotations were manually performed by comparison with other Xinmoviridae using BLASTx (https://blast.ncbi.nlm.nih.gov/Blast.cgi). Putative transmembrane domains were identified using TMHMM (http://www.cbs.dtu.dk/services/TMHMM/). The RNA-dependent RNA polymerase sequence was compared with other Xinmoviridae available at NCBI using ClustalW v.2.1 (8). Phylogeny was inferred by Maximum Likelihood with 1000 bootstrap replicates using IQ-TREE v2.3.6 (9), and topologies were visualized with FigTree v.1.4.4 (10). All tools were run with default parameters.
The sequencing yielded 15,694,292 reads of which 14,536,750 remained after quality control. The recovered contig was 12,150 bp long, with 24,834 mapped reads, GC content of 42.52%, and an average coverage of 272×. This contig contained five ORFs organized similarly to those observed in the Mononegavirales, in the following 5′−3′ terminal gene order: nucleoprotein (NP; 1,311 nt), small transmembrane protein (STM; 312 nt), hypothetical protein (1,317 nt), glycoprotein (G; 2,004 nt), and RNA-dependent RNA polymerase (RdRp; 6,057 nt) (Fig. 1A). The UTR (5′−3′) region was not determined due to low coverage (Fig. 1B). The RdRp sequence was closely related to the Albipes mosquito Gordis-like virus (GenBank accession XGU11772), sharing 72.83% (amino acids) and 65.89% (nucleotides) identity. Phylogenetic analysis showed that the obtained sequence clustered with other members of the Gordisvirus genus, with 100% bootstrap support (Fig. 1C). According to ICTV, an amino acid identity of <66% in the RdRp region is a criterion for species demarcation within the Gordisvirus genus (1). Therefore, we propose that the recovered genome could be classified as a divergent member of the genus Gordisvirus, tentatively named “Mosquito Albipes Gordis-like strain 1”.
(A) Genome organization of Albipes mosquito Gordis-like virus strain 1. Predicted open reading frames are indicated by arrows. (B) Genome coverage is represented by the blue graph. The untranslated terminal regions (UTRs) were not determined due to low read coverage in these regions. (C) Phylogenetic relationship between the Albipes mosquito Gordis-like virus strain 1 RdRp sequence (highlighted in red) and sequences of viruses from the Xinmoviridae family available in the NCBI database. The phylogenetic tree was inferred using the maximum likelihood method with the Q.pfam +F + R6 substitution model for amino acids and statistical support of bootstrap with 1000 replicates. The tree was rooted using the midpoint method implemented in FigTree v.1.4.4. The scale bar indicates amino acid substitutions per site. Bootstrap support values are shown at each node, and GenBank accession numbers are listed to the left of the virus names.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Sharpe S, Paraskevopoulou S. 2023. ICTV Virus Taxonomy profile: Xinmoviridae 2023. J Gen Virol 104:001906. doi:10.1099/jgv.0.001906 PMC 1264309537882775 · doi ↗ · pubmed ↗
- 2Aragão CF, da Silva SP, do Nascimento BLS, da Silva FS, Nunes Neto JP, Pinheiro VCS, Cruz ACR. 2023. Shotgun metagenomic sequencing reveals virome composition of mosquitoes from a transition ecosystem of North-Northeast Brazil. Genes (Basel) 14:1443. doi:10.3390/genes 1407144337510347 PMC 10379392 · doi ↗ · pubmed ↗
- 3Maia LJ, Silva AB, Oliveira C de, Campos FS, Silva L da, Abreu F de, Ribeiro BM. 2024. Sylvatic mosquito viromes in the Cerrado biome of Minas Gerais, Brazil: discovery of new viruses and implications for arbovirus transmission. Viruses 16:1276. doi:10.3390/v 1608127639205250 PMC 11359572 · doi ↗ · pubmed ↗
- 4Chen S, Zhou Y, Chen Y, Gu J. 2018. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34:i 884–i 890. doi:10.1093/bioinformatics/bty 56030423086 PMC 6129281 · doi ↗ · pubmed ↗
- 5Li D, Liu CM, Luo R, Sadakane K, Lam TW. 2015. MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 31:1674–1676. doi:10.1093/bioinformatics/btv 03325609793 · doi ↗ · pubmed ↗
- 6Buchfink B, Xie C, Huson DH. 2015. Fast and sensitive protein alignment using DIAMOND. Nat Methods 12:59–60. doi:10.1038/nmeth.317625402007 · doi ↗ · pubmed ↗
- 7Huson DH, Auch AF, Qi J, Schuster SC. 2007. MEGAN analysis of metagenomic data. Genome Res 17:377–386. doi:10.1101/gr.596910717255551 PMC 1800929 · doi ↗ · pubmed ↗