Porphyria Cutanea Tarda: A Phenotypic Expression of Several Genes
Sebastián J Vázquez-Folch, Gabriel A Jimenez-Berrios, Natalio Izquierdo, Victor Vazquez

TL;DR
This paper discusses three porphyria cases in Puerto Rico, emphasizing the role of genetic testing in diagnosis and highlighting the genetic diversity of the condition.
Contribution
The study reports a previously undocumented PCT case with elevated uroporphyrin but no genetic mutations, suggesting possible unknown genetic factors.
Findings
A PCT case showed elevated uroporphyrin levels without detectable genetic mutations.
Genetic testing identified FECH and HFE gene mutations in EPP and PCT cases.
The study underscores the need for further research into porphyria's genetic and phenotypic diversity.
Abstract
Porphyria comprises a group of rare inherited or acquired disorders characterized by defects in the heme biosynthetic pathway, resulting in the accumulation of porphyrins or their precursors. This study presents three cases of porphyria in Puerto Rico, including erythropoietic protoporphyria (EPP) and porphyria cutanea tarda (PCT). Genetic testing revealed a heterozygous mutation in the FECH gene in the EPP case and an HFE gene mutation in a PCT case with hereditary hemochromatosis. A previously undocumented case of PCT with elevated uroporphyrin levels but negative genetic panel results raises questions about the genetic basis of porphyria. Our findings highlight the importance of genetic testing in diagnosing and managing porphyria, emphasizing the need for further research into its genetic and phenotypic diversity. This study contributes to the understanding of porphyria in Puerto…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
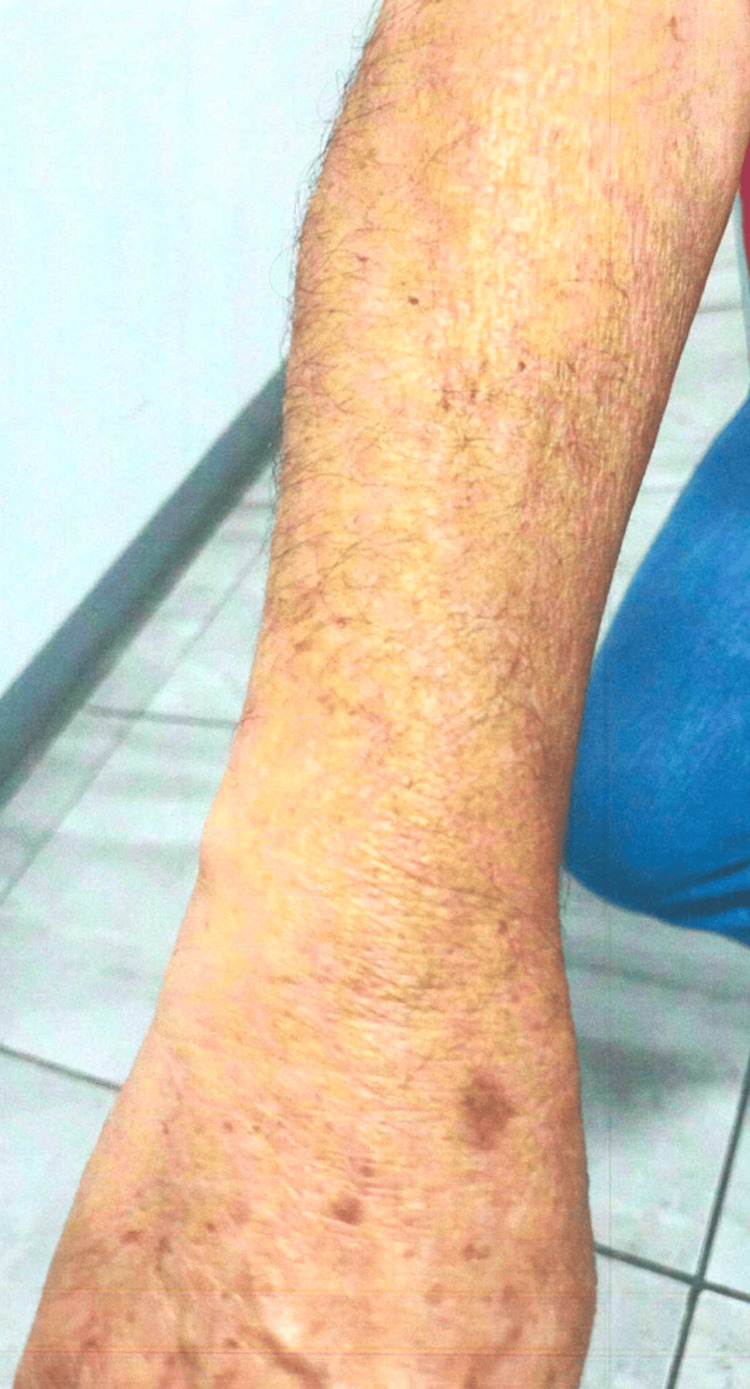 Figure 1
Figure 1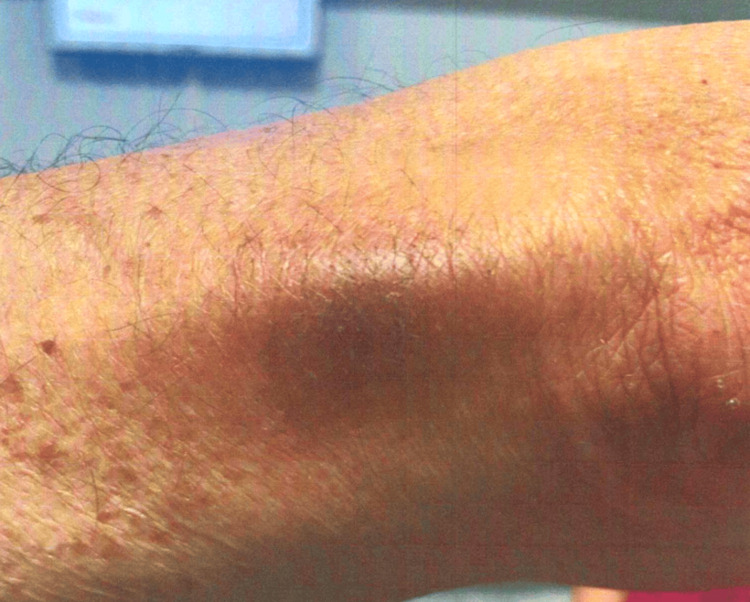 Figure 2
Figure 2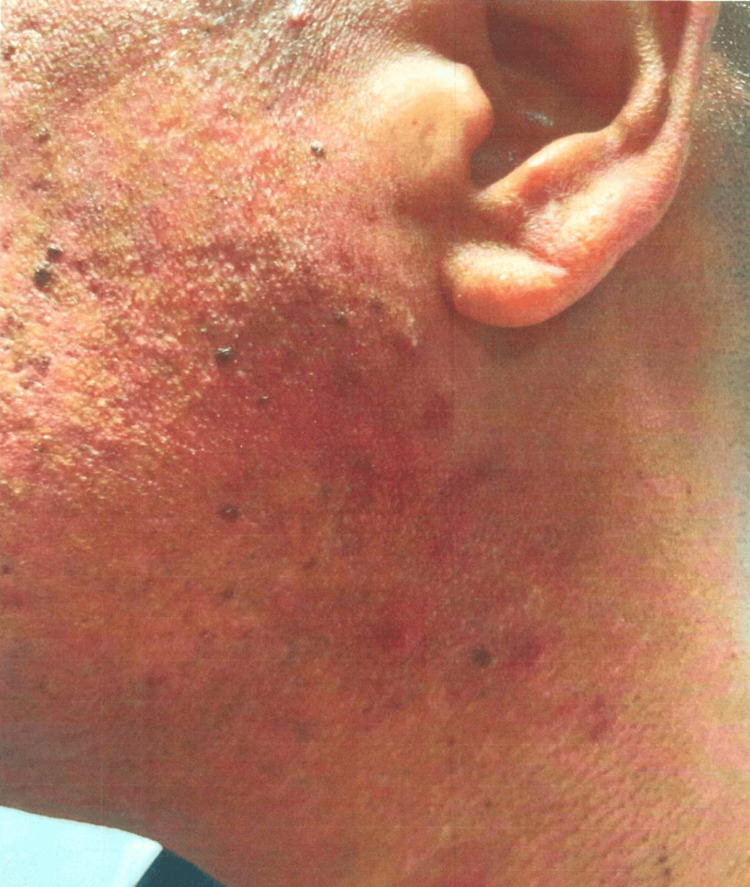 Figure 3
Figure 3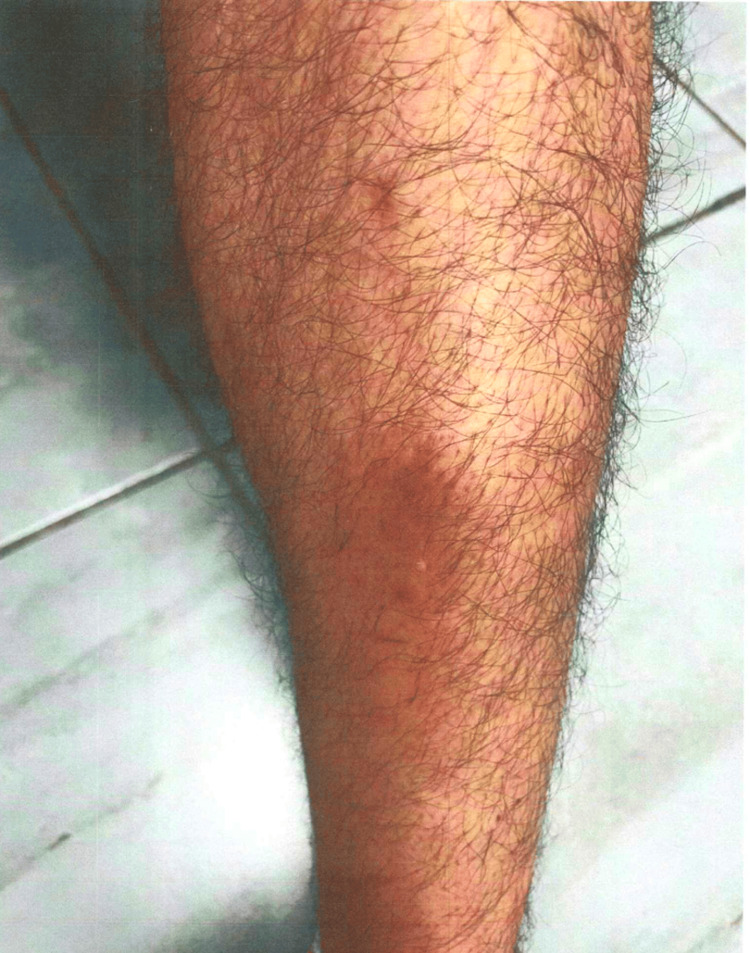 Figure 4
Figure 4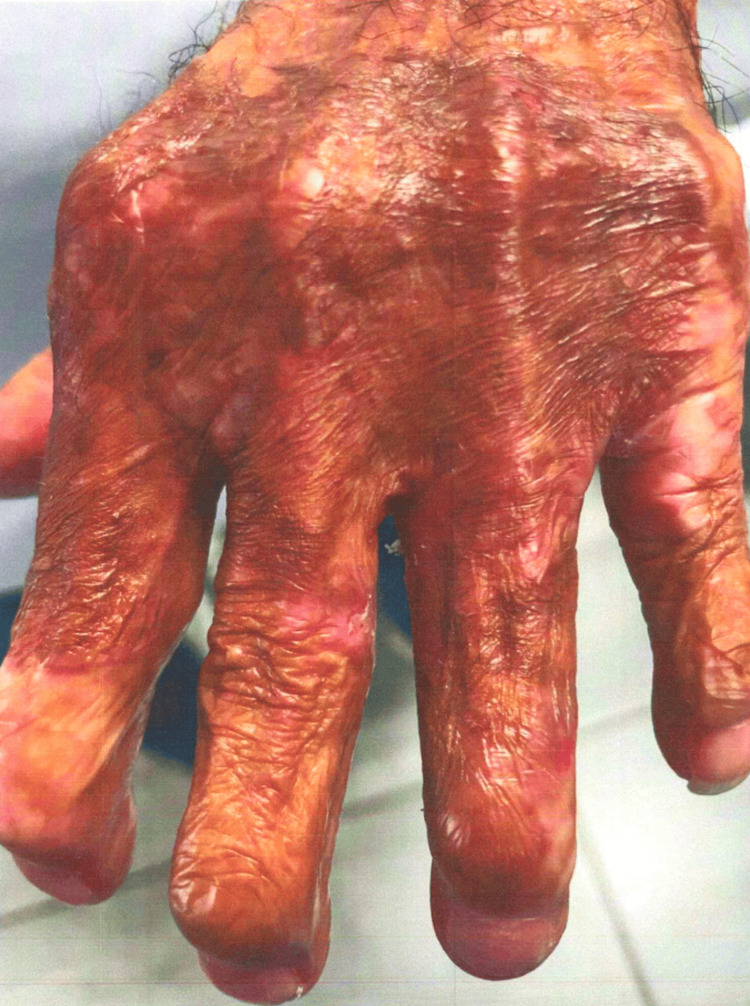 Figure 5
Figure 5 Figure 6
Figure 6| Case number | Age/sex | Type of porphyria | Gene | Mutation | Organ involvement | Initial treatment | Response to initial treatment |
| 1 | 68/F | Erythropoietic protoporphyria | FECH gene | (c.315-48T>C) | Skin | Phlebotomy | Responsive |
| 2 | 41/M | Inconclusive | Inconclusive | Inconclusive | Skin | Phlebotomy | Developed intolerance; consider hydroxychloroquine |
| 3 | 70/M | Porphyria cutanea tarda | HFE gene | C282Y (c.845G>A, p.Cys282Tyr) and H63D (C.187C>G, p.His63Asp) | Skin | Phlebotomy | Responsive |
| Laboratory test results pre-phlebotomy | |
| Uroporphyrins (UP) | 5 ug/L |
| Heptacarboxyl (7-CP) | 1 μg/L |
| Hexacarboxyl (6-CP) | 43 μg/L |
| Hexacarboxyl (6-CP), 24 hr | 75 μg/24 hr (HIGH) |
| Pentacarboxyl (5-CP) | <1 μg/L |
| Coproporphyrin (CP) I | 8 μg/L |
| Coproporphyrin (CP) III | 33 μg/L |
| Laboratory test results post-phlebotomy | |
| Uroporphyrins (UP) | 6 μg/L |
| Heptacarboxyl (7-CP) | <1 μg/L |
| Hexacarboxyl (6-CP) | <1 μg/L |
| Pentacarboxyl (5-CP) | 4 μg/L |
| Pentacarboxyl (5-CP), 24 hr | 10 μg/24 hr (HIGH) |
| Coproporphyrin (CP) I | 6 μg/L |
| Coproporphyrin (CP) III | 13 μg/L |
| Laboratory test results pre-phlebotomy | |
| Uroporphyrins (UP) | 21 μg/L (HIGH) |
| Heptacarboxyl (7-CP) | 1 μg/L |
| Hexacarboxyl (6-CP) | 1 μg/L |
| Pentacarboxyl (5-CP) | 2 μg/L |
| Coproporphyrin (CP) I | 11 μg/L |
| Coproporphyrin (CP) III | 6 μg/L |
| Laboratory test results post-phlebotomy | |
| Uroporphyrins (UP) | 11 μg/L |
| Heptacarboxyl (7-CP) | <1 μg/L |
| Hexacarboxyl (6-CP) | <1 μg/L |
| Pentacarboxyl (5-CP) | <1 μg/L |
| Coproporphyrin (CP) I | 13 μg/L |
| Coproporphyrin (CP) III | 29 μg/L |
| Laboratory test results pre-phlebotomy | |
| Uroporphyrins (UP) | 40 μg/L (HIGH) |
| Heptacarboxyl (7-CP) | 8 μg/L (HIGH) |
| Hexacarboxyl (6-CP) | <1 μg/L |
| Pentacarboxyl (5-CP) | <1 μg/L |
| Coproporphyrin (CP) I | 43 μg/L (HIGH) |
| Coproporphyrin (CP) III | 5 μg/L |
| Laboratory test results post-phlebotomy | |
| Uroporphyrins (UP) | 33 μg/L (HIGH) |
| Heptacarboxyl (7-CP) | <1 μg/L |
| Hexacarboxyl (6-CP) | <1 μg/L |
| Pentacarboxyl (5-CP) | <1 μg/L |
| Coproporphyrin (CP) I | 26 μg/L (HIGH) |
| Coproporphyrin (CP) III | 52 μg/L (HIGH) |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsPorphyrin Metabolism and Disorders · Heme Oxygenase-1 and Carbon Monoxide · Methemoglobinemia and Tumor Lysis Syndrome
Introduction
Porphyria is a group of rare inherited or acquired disorders resulting from the malfunction of the heme biosynthetic pathway, leading to the accumulation of porphyrins or their precursors [1,2]. The heme biosynthetic pathway is crucial for the production of heme, an essential component of hemoglobin, myoglobin, and various cytochromes [3]. Defects in this pathway may result in a variety of clinical manifestations, including the skin and nervous system [4].
Porphyrias are classified and include erythropoietic protoporphyria (EPP), porphyria cutanea tarda (PCT), acute intermittent porphyria, hereditary coproporphyria, variegate porphyria, and congenital erythropoietic porphyria [5].
EPP is primarily caused by autosomal recessive mutations in the FECH gene, which encodes the enzyme ferrochelatase [6]. This enzyme is responsible for the last step in heme production, where iron is inserted into protoporphyrin IX to form heme [7]. A deficiency in ferrochelatase leads to the accumulation of protoporphyrin IX in erythrocytes, plasma, and tissues. Patients with EPP typically have photosensitivity, experiencing burning pain and erythema upon exposure to sunlight. Upon chronicity, the disease may lead to liver complications due to the buildup of protoporphyrins in the bile [6].
PCT is the most common type of porphyria and is usually acquired, although it may also have a genetic inheritance. PCT is characterized by a deficiency in the enzyme uroporphyrinogen decarboxylase (UROD), which leads to the accumulation of uroporphyrins in the liver, plasma, and skin [8]. This accumulation causes photosensitivity, resulting in painful blistering, fragility, and hyperpigmentation of sun-exposed skin areas [9]. PCT can be associated with liver dysfunction and iron overload, and it is often linked with genetic variants such as mutations in the HFE gene, leading to hereditary hemochromatosis [10]. An increased frequency of hereditary hemochromatosis gene mutations occurs in patients with PCT [11].
Case presentation
The key findings of the patients are summarized in Table 1.
Case 1: EPP
A 68-year-old female patient had skin lesions in sunlight-exposed areas and pruritus urticaria and was diagnosed with porphyria in July 2010. She was referred by her dermatologist for therapeutic phlebotomy. Her laboratory results showed elevated erythropoietin levels at 43.65 mIU/mL. Upon genetic testing (Invitae, San Francisco, California, United States), the patient had a heterozygous mutation in the FECH gene (c.315-48T>C). FibroScan (Echosens, Paris, France) was completed in December 2023. The liver stiffness measurement (LSM) was 5.2 kPa. The patient was diagnosed with minimal hepatic fibrosis. Laboratory test results are shown in Tables 2-3.
The patient's dermatologic findings are shown in Figures 1-2.
Case 1: patient's dermatologic findings in the right arm
Case 1: patient's dermatologic findings in the left upper extremity
Case 2: PCT
Case 1's offspring, a 41-year-old male patient, had a history of skin blistering and photosensitivity. He was diagnosed with PCT in 2010 and was referred by his dermatologist for phlebotomy, which lowered his hemoglobin to 10 g/dL. His symptoms improved post-phlebotomy. His laboratory results in 2010 showed elevated levels of various porphyrins, including uroporphyrins and coproporphyrins. His erythropoietin levels are high, 19.37 mIU/mL, in 2024. Genetic studies (Invitae) showed no mutations in the porphyria panel. The patient tested negative for the following porphyria enzymes: ALAD, ALAS2, CPLX, CPOX, FECH, GATA1, HMBS, PPOX, UROD, and UROS. He developed intolerance to phlebotomy due to dizziness, tachycardia, and hypotension. Therefore, the patient was advised to follow up with quantitative porphyrins and consider starting low doses of hydroxychloroquine (Plaquenil) for symptom management. Laboratory test results are shown in Tables 4-5.
The patient's dermatologic findings are shown in Figures 3-4.
Case 2: patient's dermatologic findings in the left side of his face/neck
Case 2: patient's dermatologic findings in the left lower extremity
Case 3: unrelated patient with PCT and hemochromatosis
A 70-year-old male patient (unrelated to the other two cases) was diagnosed with PCT. Upon genetic testing, the patient had two pathogenic variants in the HFE gene, C282Y (c.845G>A, p.Cys282Tyr) and H63D (C.187C>G, p.His63Asp), which are commonly associated with hereditary hemochromatosis. This patient, an HFE gene mutation carrier, had symptoms of PCT, which were managed with therapeutic phlebotomy. Laboratory test results are shown in Tables 6-7.
The patient's dermatologic findings are shown in Figures 5-6.
Case 3: patient's dermatologic findings on his left hand
Case 3: patient's dermatologic findings in the left side of his neck
Discussion
Porphyria is a group of diseases that are characterized by abnormalities in the heme biosynthetic pathway, leading to the accumulation of porphyrins or their precursors, which can cause a variety of symptoms, including photosensitivity, skin lesions, and neurological complications.
Several genetic mutations can lead to porphyria: EPP, PCT, acute intermittent porphyria, hereditary coproporphyria, variegate porphyria, and congenital erythropoietic porphyria [5].
EPP is most commonly caused by autosomal recessive mutations in the FECH gene, leading to the accumulation of protoporphyrins in the skin. This leads to painful photosensitivity and potential liver damage due to the biliary excretion of protoporphyrins [12]. Case 1 was clinically diagnosed with porphyria, and therapeutic phlebotomy was done to manage the patient's signs and symptoms. Upon genetic analysis, Case 1 had a heterozygous mutation in the FECH gene. This is compatible with a partial ferrochelatase activity and, thus, a less severe clinical presentation of the disease.
Furthermore, Case 2 (son of Case 1) was previously diagnosed with PCT. Photosensitivity, a symptom of congenital erythropoietic porphyria, can also occur in the cutanea tarda and "mixed" hepatic types [13]. Genetic testing showed that the patient had no mutations in the porphyria genes. Although the patient had similar skin blistering as Case 1, the genetic findings remain inconclusive.
Case 2 presented with solar urticaria and other acute skin changes caused by ultraviolet radiation. Additionally, the patient's uroporphyrin levels were elevated at the time of collection in 2010. These findings raise the question of whether the patient can be diagnosed with porphyria, despite a negative result on the porphyria genetic panel. To our knowledge, these findings have not been previously described.
Hereditary hemochromatosis, often associated with PCT, is an autosomal recessive condition that leads to iron overload, further complicating the clinical management of porphyria [11,14]. Case 3 was diagnosed with PCT. The genetic assay detected two pathogenic variants in the HFE gene: C282Y and H63D. Both variants are commonly associated with hereditary hemochromatosis, which is characterized by a disorder in iron metabolism. Li and co-workers report a rare case of hereditary hemochromatosis with similar pathogenic variants in the HFE gene [15]. After regular phlebotomy, the patient's serum ferritin (SF), serum iron (SI), and transferrin saturation (TSAT) decreased. Additionally, transferrin (TF) and liver function returned to normal, and the patient's signs and symptoms improved. For our patient, therapeutic phlebotomy was performed. Post-phlebotomy, TF level was 336 mg/dL, SI 18 mg/dL, iron 30 mg/dL, iron saturation 6.9%, hemoglobin 16.9 g/dL, and hematocrit 53.6%. Case 3 of this report underscores the importance of considering genetic testing and managing iron levels in patients with PCT.
Overall, these cases illustrate the diverse presentations and complexities involved in the diagnosis and management of the porphyrias. Understanding the underlying genetic mutations and their biochemical consequences is crucial for the effective co-management, treatment, and improvement of patient outcomes.
Genetic studies in patients with porphyria are warranted. Genetic testing may provide critical insights into the diagnosis, management, and prognosis of various hematologic hereditary diseases. This is the first report of the genetics of porphyria in Puerto Rico.
Limitations of the study include the small number of PCT cases. Further studies will continue to elucidate the genetics leading to PCT in patients, enhancing our understanding and treatment of this complex group of disorders.
Conclusions
We report on the clinical and genetic findings of patients with porphyria. With case presentations of EPP and PCT, we underscore the complexities in diagnosing and managing these disorders, particularly in the context of genetic variability. The identification of a novel case in Puerto Rico enriches the understanding of porphyria's phenotypic expression in diverse populations. Our findings emphasize the critical role of genetic testing in elucidating the etiology of porphyria, guiding therapeutic interventions, and improving patient outcomes. Further studies should continue to explore the genetic and environmental factors contributing to porphyria, facilitating more precise diagnostic and management strategies.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Exploring current and emerging therapies for porphyrias Liver Int JericóD Córdoba KM Urigo F 217421904420243881395310.1111/liv.15979 · doi ↗ · pubmed ↗
- 2Heme in pathophysiology: a matter of scavenging, metabolism and trafficking across cell membranes Front Pharmacol Chiabrando D Vinchi F Fiorito V Mercurio S Tolosano E 61520142478276910.3389/fphar.2014.00061 PMC 3986552 · doi ↗ · pubmed ↗
- 3Cell biology of heme Am J Med Sci Ponka P 2412563181999 https://pubmed.ncbi.nlm.nih.gov/10522552/1052255210.1097/00000441-199910000-00004 · doi ↗ · pubmed ↗
- 4Acute hepatic porphyria Stat Pearls [Internet] Kothadia JP La Freniere K Shah JM Treasure Island (FL)Stat Pearls Publishing 2023 https://pubmed.ncbi.nlm.nih.gov/30725863/30725863 · pubmed ↗
- 5Heme biosynthesis and the porphyrias Mol Genet Metab Phillips JD 16417712820193132628710.1016/j.ymgme.2019.04.008PMC 7252266 · doi ↗ · pubmed ↗
- 6Updates on the diagnosis and management of the most common hereditary porphyrias: AIP and EPP Hematology Am Soc Hematol Educ Program Linenberger M Fertrin KY 400410202020203327567710.1182/hematology.2020000124 PMC 7727547 · doi ↗ · pubmed ↗
- 7Clinical and molecular epidemiology of erythropoietic protoporphyria in Italy Eur J Dermatol Ventura P Brancaleoni V Di Pierro E 5325403020203302147310.1684/ejd.2020.3880 · doi ↗ · pubmed ↗
- 8The clinical management of porphyria cutanea tarda: an update Liver Int Sarkany RP Phillips JD 219121964420243881394910.1111/liv.15980 · doi ↗ · pubmed ↗