Linking Fluorine with Bio-Derived Furfural: Aiming Towards More Sustainable Fluorinated Polymers and Drugs
Konstantin I. Galkin, Irina V. Sandulenko

TL;DR
This paper explores how fluorine can be added to bio-based furfural to create more sustainable polymers and drugs.
Contribution
The paper introduces new methods for incorporating fluorine into furfural-derived compounds for sustainable applications.
Findings
Fluorinated furans can be synthesized from bio-derived furfural.
These fluorinated compounds show promise in pharmaceuticals and materials science.
Current trends suggest a growing interest in sustainable fluorinated products.
Abstract
This perspective highlights current trends and recent advances in the introduction of fluorine and fluoroalkyl moieties into the furanic core of biobased furfural-derived furans. Existing and potential applications of these fluorinated building blocks in the development of pharmaceuticals and advanced materials are also discussed.
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
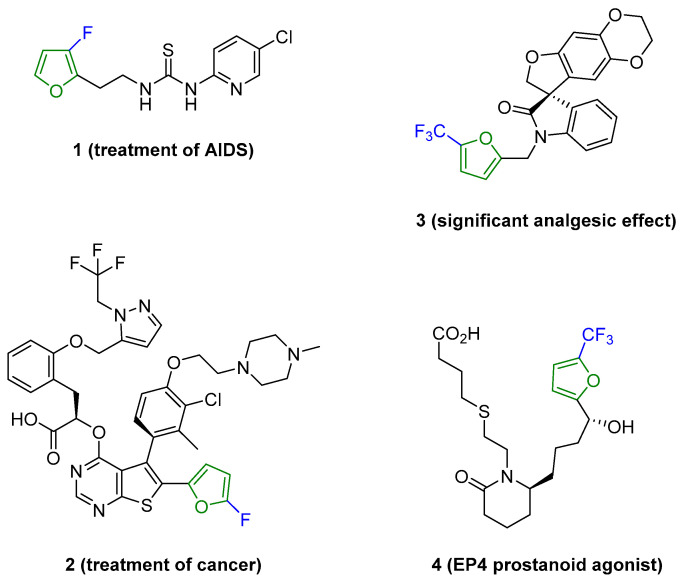 Figure 1
Figure 1 Figure 2
Figure 2 Figure 3
Figure 3 Figure 4
Figure 4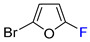 Figure 5
Figure 5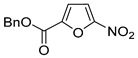 Figure 6
Figure 6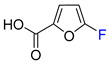 Figure 7
Figure 7 Figure 8
Figure 8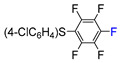 Figure 9
Figure 9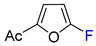 Figure 10
Figure 10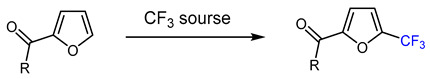 Figure 11
Figure 11 Figure 12
Figure 12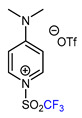 Figure 13
Figure 13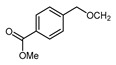 Figure 14
Figure 14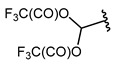 Figure 15
Figure 15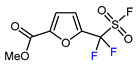 Figure 16
Figure 16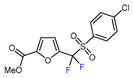 Figure 17
Figure 17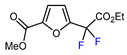 Figure 18
Figure 18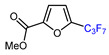 Figure 19
Figure 19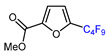 Figure 20
Figure 20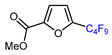 Figure 21
Figure 21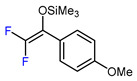 Figure 22
Figure 22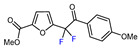 Figure 23
Figure 23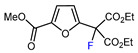 Figure 24
Figure 24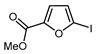 Figure 25
Figure 25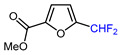 Figure 26
Figure 26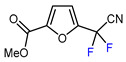 Figure 27
Figure 27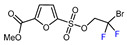 Figure 28
Figure 28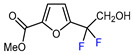 Figure 29
Figure 29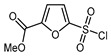 Figure 30
Figure 30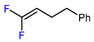 Figure 31
Figure 31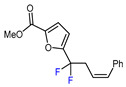 Figure 32
Figure 32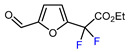 Figure 33
Figure 33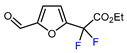 Figure 34
Figure 34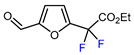 Figure 35
Figure 35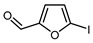 Figure 36
Figure 36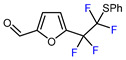 Figure 37
Figure 37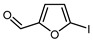 Figure 38
Figure 38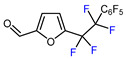 Figure 39
Figure 39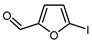 Figure 40
Figure 40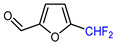 Figure 41
Figure 41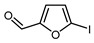 Figure 42
Figure 42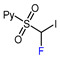 Figure 43
Figure 43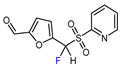 Figure 44
Figure 44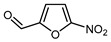 Figure 45
Figure 45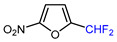 Figure 46
Figure 46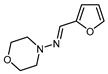 Figure 47
Figure 47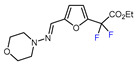 Figure 48
Figure 48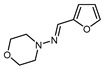 Figure 49
Figure 49 Figure 50
Figure 50|
| ||||
|
|
|
|
|
|
| 1 | Me | CF3COOH, (4-ClPh)2SO (2 eq.), Ru(bpy)3Cl2 (1 mol. %), DCE, RT, 427 nm LED | 62 | [ |
| 2 | Me | TFAA, Me2C=NOH (2 eq.), Ru(bpy)3Cl2, DCE, RT, 390 nm LED | 62 | [ |
| 3 | OMe | CF3COOH, (4-ClPh)2SO (2 eq.), Ru(bpy)3Cl2 (1 mol. %), DCE, RT, 427 nm LED | 59 | [ |
| 4 | OMe | TFAA, Me2C=NOH (2 eq.), Ru(bpy)3Cl2, DCE, RT, 390 nm LED | 74 | [ |
| 5 | OMe | TFAA, Ir(ppy)3 (3 mol. %), EtOAc, 40 °C, 30 W blue LED | 64 | [ |
| 6 | OMe | C6F5I(OCOCF3)2, Ru(bpy)3(PF6)2 (2 mol. %), CH3CN, 35 °C, blue LED | 41 | [ |
| 7 | OMe | (CF3SO2)2O, Ru(bpy)3Cl2 (2 mol. %), pyridine, DCE, RT, LED | 74 | [ |
| 8 | OMe | CF3SO2Na, [Ru(bpy)3][PF6]2, LiClO4, CH3CN, RT, blue LED, CCE at 4.0 mA | 77 | [ |
| 9 | OMe | CF3SO2Cl, K2HPO4, 100 Hz, (+)C/(−)C, 4.8 V, CH3CN, 0.125 M LiClO4 | 32 | [ |
| 10 | OC8H17 | CF3COOH, (4-ClPh)2SO (2 eq.), Ru(bpy)3Cl2 (1 mol. %), DCE, RT, 427 nm LED | 58 | [ |
| 11 | OC8H17 | TFAA, Me2C=NOH (2 eq.), Ru(bpy)3Cl2, DCE, RT, 390 nm LED | 60 | [ |
| 12 | NHMe | CF3SO2Na, [Ru(bpy)3][PF6]2, LiClO4, CH3CN, RT, blue LED, CCE at 4.0 mA | 69 | [ |
| 13 | NH(CH2COOEt) | CF3SO2Na, [Ru(bpy)3][PF6]2, LiClO4, CH3CN, RT, blue LED, CCE at 4.0 mA | 70 | [ |
| 14 | OH | TFAA, Me2C=NOH (2 eq.), Ru(bpy)3Cl2, DCE, RT, 390 nm LED | 68 | [ |
| 15 | OH | (CF3SO2)2O, Ru(bpy)3Cl2 (2 mol. %), pyridine, DCE, RT, LED | 72 | [ |
|
| ||||
|
|
|
|
|
|
| 1 | CH3 | CF3SO2Cl, Ru-cat., Mg acetate, CH3CN, RT, blue light | 86 | [ |
| 2 | CH3 | CF3SO2Cl, Ru(phen)3Cl2 (1 mol. %), K2HPO4, CH3CN, RT, 26 W light | 87 | [ |
| 3 | CH3 | Co(III)-CF3 complex, NMP, RT, 16 W light | 87 | [ |
| 4 | CH3 | CF3I, Ru(bpy)3Cl2 (1 mol. %), TMEDA, CH3CN, blue LED | 75 | [ |
| 5 | CH3 | TMSCF3 (2eq.), PhI(OAc) (2 eq.), AgF (25 mol. %), DMSO, RT | 51 | [ |
| 6 | CH3 | 63 | [ | |
| 7 | CH3 | CF3SO2Cl (2 eq.), K2HPO4, 100 Hz, (+)C/(−)C, CH3CN, 0.125 M LiClO4 | 43 | [ |
| 8 | CH2S(CHO) | CF3SO2Cl, CdSe (10 mol. %), K2HPO4, CH3CN, RT, blue LED | 68 | [ |
| 9 |
| TFAA, urea-hydrogen peroxide, CH2Cl2, 0 °C | 49 | [ |
| 10 | CH2OBn | (C2F5CO)2O, urea-hydrogen peroxide, CH2Cl2, 0 °C | 46 | [ |
| 11 |
| CF3COOH, XeF2, CH2Cl2, 0 → 20 °C, then H2O, CF3COOH | 30 1 | [ |
| № | Furan | Reaction Conditions | Product | Yield | Ref. |
|---|---|---|---|---|---|
| 1 | Methyl 2-furoate | ICF2SO2F, [Ir(dFCF3ppy)2(bpy)]PF6 (1 mol. %), AgOTf (1 eq.), DMC, RT, blue LED |
| 80 | [ |
| 2 | Methyl 2-furoate | BrCF2SO2(4-Cl-Ph), Mn2(CO)10 (10 mol. %), Davephos (15 mol. %), K2CO3, DCM, white LED |
| 86 | [ |
| 3 | Methyl 2-furoate | BrCF2CO2Et, Pd(PPh3)4 (5 mol. %), Xantphos (10 mol. %), K2CO3, dioxane, Ar, 110 °C |
| 67 | [ |
| 4 | Methyl 2-furoate | C3F7,XeF2, CH2Cl2, RT |
| 33 | [ |
| 5 | Methyl 2-furoate | IC4F9 (30 eq.), CuI (10 mol. %), phen (20 mol. %), 2,4,6-collidine, CH2Cl2, 110 °C |
| 46 | [ |
| 6 | Methyl 2-furoate | IC4F9, [Cu(bcp)DPEphos]PF6 (0.1 eq.), KOAc, CH2Cl2, blue LED |
| 51 | [ |
| 7 | Methyl 2-furoate |
| 61 | [ | |
| 8 | Methyl 2-furoate | BrCF(CO2Et)2, |
| 72 | [ |
| 9 |
| [Ph4P]+[Cu(CF2H)2]– (1 eq.), DMAc, 90 °C |
| 56 | [ |
| 10 | Ethyl 2-furoate | BrCF2CN, [Ir(dtbbpy)(ppy)2][PF6], DMF, blue LED |
| 74 | [ |
| 11 |
| [Ru(bpy)3]Cl2 6H2O (0.1 mol. %), NBu3 (1.5 eq.), HCOOH (1.5 eq.), DMSO, RT, blue LED |
| 73 | [ |
| 12 |
|
| 50 | [ |
| № | Furan | Reaction Conditions | Product | Yield | Ref. |
|---|---|---|---|---|---|
| 1 | Furfural | BrCF2CO2Et, CuI (10 mol. %), PMDETA (1.5 eq.), CH3CN, 80 °C |
| 50 | [ |
| 2 | Furfural | BrCF2CO2Et, Ir(ppy)3 (0.5 mol. %), phen (20 mol. %), NaOAc (2 eq.), CH3CN, RT, 3 W blue LED |
| 44 | [ |
| 3 | Furfural | BrCF2CO2Et, [Ru( |
| 62 | [ |
| 4 |
| TMSCF2Br (7.2 eq.), CuBr (5.2 eq.), ArSH (12 eq.), phen, 18-crown-6, NaH, C6F6, NMP, 60 °C |
| 81 | [ |
| 5 |
| TMSCF3 (1.8 eq.), C6F5TMS, CuCl (5.2 eq.), KF (12 eq.), 60 °C |
| 75 | [ |
| 6 |
| (DMPU)2Zn(CHF2)2, (dppf)Ni(COD) (15 mol. %), DMSO, RT |
| 78 | [ |
| 7 |
|
| 67 | [ | |
| 8 |
| Deoxo-Fluor (1.5 eq.) CH2Cl2, RT |
| 68 | [ |
| 9 |
| BrCF2CO2Et, CuBr2 (10 mol. %), DTBDPy (10 mol. %), B2pin2 (30 mol. %), NaHCO3, dioxane, 80 °C |
| 51 | [ |
| 10 |
|
| 69 | [ | |
| 11 | 2-Acetylfuran | BrCF2CO2Et, CuI (10 mol. %), PMDETA (1.5 eq.), CH3CN, 80 °C |
| 53 | [ |
| 12 | 2-Acetylfuran | BrCF(CO2Et)2, |
| 65 | [ |
| 13 | 2-Furonitrile | ICF2SO2F, [Ir(dFCF3ppy)2(bpy)]PF6 (1 mol. %), AgOTf (1 eq.), DMC, RT, blue LED |
| 85 | [ |
| № | Furan | Reaction Conditions | Product | Yield | Ref. |
|---|---|---|---|---|---|
| 1 | 2-Methylfuran | BrCF2CO2Et, CuI (10 mol. %), K2CO3, DMF, 60 °C |
| 61 | [ |
| 2 | 2-Methylfuran | BrCF2CO2Et, Pd(PPh3)4 (5 mol. %), Xantphos (10 mol. %), K2CO3, dioxane, Ar, 110 °C |
| 57 | [ |
| 3 | 2-Ethylfuran | BrCF2CO2Et, |
| 83 | [ |
| 4 | 2-Ethylfuran | PhSO2CF2I, Pd2(dba)3 (5 mol. %), Xantphos (20 mol. %), Cs2CO3, CHCl3, 60 °C |
| 96 | [ |
| 5 | 2-Ethylfuran | PhSO2CF2I, Ru(bpy)3Cl2•6H2O (1 mol. %), K2HPO4, CH2Cl2, 40 °C, 26W light |
| 96 | [ |
| 6 | 2-Ethylfuran | BrCF2CO2Et, Pd(PPh3)4 (5 mol. %), Xantphos (10 mol. %), K2CO3, dioxane, Ar, 110 °C |
| 63 | [ |
| 7 |
| PhSO2CF2I, Ru(bpy)3Cl2•6H2O (1 mol. %), K2HPO4, CH2Cl2, 26 W light, 40 °C |
| 90 | [ |
| 8 |
| BrCF2CO2Et, Pd(PPh3)4 (5 mol. %), Xantphos (10 mol. %), K2CO3, dioxane, Ar, 110 °C |
| 80 | [ |
| 9 | Furfuryl alcohol | IC4F9 (30 eq.), CuI (10 mol. %), 1,10 phenantroline (20 mol. %), 2,4,6-collidine, CH2Cl2, 110 °C |
| 74 | [ |
| 10 |
| BrCF2CO2Et, CuI (10 mol. %), K2CO3, DMF, 80 °C |
| 64 | [ |
| 11 |
| BrCF2CO2Et, CuI (10 mol. %), K2CO3, DMF, 80 °C |
| 65 | [ |
| 12 |
| BrCF2CO2Et, CuI (10 mol. %), K2CO3, DMF, 80 °C |
| 63 | [ |
| 13 |
| DAST (3.5 eq.), CH2Cl2, RT |
| 60 | [ |
- —Russian Science Foundation
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsFluorine in Organic Chemistry · Catalysis for Biomass Conversion · Chemical Synthesis and Reactions
1. Introduction
While naturally occurring organofluorine compounds are rare, [1] these synthetic products have a significant impact on academic and industrial fields. Fluorine is the most electronegative element that is sterically similar to hydrogen [2]; therefore, carbon–fluorine bonds are characterized by high stability, low polarization and a strong electron-withdrawing ability [3]. Consequently, the introduction of a fluorine atom does not substantially increase the volume of the molecule, yet it often leads to significant changes in physical, chemical, stereochemical, physicochemical, and biological properties of small organic molecules and polymers [4,5,6,7,8].
Due to their unique advantages, fluorinated compounds are widely used, particularly in the development of advanced pharmaceuticals, agrochemicals, and materials [9,10,11,12]. The importance of fluorine in the development of pharmaceuticals and agrochemicals is mostly a consequence of the considerable influence of fluorination on bioactivity profiles, metabolic stability, and physicochemical properties such as acidity/basicity, lipophilicity, and solubility [2,5,13,14,15,16,17]. Additionally, fluorine atoms can participate in hydrogen bonding as electron pair donors, thereby stabilizing certain conformations [18]. These advantages of fluorination contribute to fluorine-containing compounds accounting for approximately 20–25% of approved small molecule pharmaceuticals, with 30–40% of agrochemicals containing at least one fluorine atom in their structure [19,20,21,22,23,24,25]. Numerous fluorinated derivatives with notable biological activity have been synthesized, including indoles [26,27], quinazolines [28,29], steroids [30], and morphinans [31,32,33].
Notably, the trifluoromethyl group is the most commonly used fluorinated group [34]. In biological studies and materials, it is particularly attractive due to the greater chemical stability of trifluoromethylated products compared to difluoromethyl- and monofluoromethyl-containing analogues, as well as its relatively low toxicity [35]. The steric demand of a trifluoromethyl group is comparable to that of an isopropyl group, and its electronegativity is similar to that of oxygen [18]. Incorporating trifluoromethyl groups into organic compounds can significantly modify their chemical reactivity, acidity, polarity, lipophilicity, metabolic stability, and binding selectivity [34,36].
Direct trifluoromethylation with or without transition metal catalysts offers an important pathway for synthesizing fluorinated targets, but it often suffers from poor selectivity, low yields, harsh reaction conditions, and the usage of corrosive and expensive CF_3_ sources, which restricts the feasibility of this approach [37,38,39]. Over the past decade, substantial efforts have been made to address these limitations. Consequently, numerous efficient methods for the direct incorporation of trifluoromethyl groups into heterocyclic molecules have been developed, utilizing electrophilic, nucleophilic, and radical trifluoromethylating reagents [34,40,41,42]. Compared to nucleophilic and electrophilic trifluoromethylation, radical trifluoromethylation allows for the direct introduction of CF_3_ into organic molecules without the need for additional steps to prepare functionalized substrates (such as organohalides and organometallic reagents), thereby providing a more efficient route to access trifluoromethylated compounds [43,44].
Carbo- and heterocycles containing fluorine or fluoroalkyl moieties are common structural motifs of many bioactive substances [9,20,45]. On the other hand, furans are perhaps one of the most prominent classes of heteroaromatic compounds with widespread occurrence in nature [46]. Numerous bioactive natural and synthetic products such as pharmaceuticals, agrochemicals and flavouring agents contain a furan ring in their structure [47,48,49,50,51,52]. The insufficient stability of many furanic compounds significantly limits their synthetic utility [53,54,55]. The presence of strong electron-withdrawing F-containing substituents at the α-carbon (C2 or C5) positions markedly improves the furan ring stability under acidic conditions [56,57]. Consequently, fluorine-functionalized furans have emerged as particularly valuable scaffolds in fine organic synthesis and prospective biologically active compounds [56,57].
Fluorine-containing furans have been utilized in the design of drugs for treatment of various diseases [57,58,59,60,61,62,63]. For instance, β-fluorofuran derivative 1, a structural analogue of trovirdine (LY 300046), exhibits high amounts of activity against the human immunodeficiency viruses (HIV) (Figure 1) [64]. Compound 2 with a α-fluorofuran moiety is a potent and selective inhibitor of the induced myeloid leukemia cell differentiation protein MCL1 and is considered a promising candidate for targeted cancer therapy [65]. Trifluoromethylated furan subunits can often be found in novel structures for drug development and agrochemicals (examples of biologically active α-trifluoromethylated furans 3 and 4 are presented on Figure 1) [21,23,60,66]; they have also found prospective applications in various materials including liquid crystals, photoresist polymers, and self-assembling monolayers [60].
Fluorinated and fluoroalkylated furans may serve as valuable synthetic intermediates. While numerous methods for the nucleophilic or transition-metal-catalyzed defluorinative C−F bond functionalization of fluorinated heterocycles are reported [67,68,69], information regarding the application of these methods for modifying fluorine-functionalized furans is limited [70]. On the other hand, the high electronegativity of the fluorine atom can make the adjacent C–H bond in a furan ring more susceptible to nucleophilic attacks that may facilitate the transformation of fluorine-containing furans into more complex fluorinated structures [71,72]. Furthermore, fluorofurans and fluoroalkylfurans can be utilized as building blocks for synthesizing non-furanic cyclic or acyclic compounds [73].
Furfural is a key renewable furanic compound produced industrially from plant biomass [74,75,76]. Due to its availability and diverse reactivity, furfural has been selected as a platform chemical [77] and is actively used as a renewable building block for biofuels, fine chemicals, monomers, and polymers [78,79,80,81]. One of the actual and promising areas of application of furfural and other bioderived furans is the sustainable production of pharmaceuticals and other bioactive compounds [82,83].
In contrast to traditional aromatic polymers, furfural-derived polymers enable the transformation into three-dimensional dynamic thermosets by utilizing the reactivity of the furan ring in Diels–Alder reactions with maleimides [84,85]. Dynamic thermosets, also known as covalent adaptable networks (CANs), dynamers or vitrimers, combine the advantages of traditional thermosets such as heat, chemical, and creep resistance, high mechanical strength, and electrical insulation with the ability to be reprocessed like thermoplastics through the activation of dynamic covalent bonds [86,87]. Current research on furan–maleimide Diels–Alder (FMDA) thermosets focuses on developing sustainable and biobased smart materials, which combine excellent mechanical properties with effective self-healing efficiency and recycling capabilities [88,89]. The self-healing mechanism in FMDA polymers relies on the reversibility of the furan–maleimide Diels–Alder reaction, which can be initiated by various external stimuli such as temperature, light, and mechanical or magnetic forces [84,85].
The modification of furfural into fluorinated furanic building blocks presents an intriguing approach to linking the biomass conversion with the production of fluorinated fine chemicals, advanced dynamic polymers, and pharmaceuticals (Scheme 1). Application of biobased furans as starting materials makes the production of targeted fluorinated products more sustainable and environmentally friendly in agreement with modern trends towards green chemical industry. In this perspective, we analyze the current challenges and recent advancements in introducing fluorine or fluoroalkyl functionalities into the furanic core of biobased furfural and most important derivatives. We also discuss the progress in applications of these fluorinated furanic building blocks in the development of pharmaceuticals and dynamic polymers.
2. Fluorination and Fluoroalkylation of Furfural-Derived Furans
While numerous methods for the incorporation of fluorine or fluorine-containing functional groups into various aromatic and heteroaromatic structures have been reported, [10,34,41,90,91,92], the functionalization of furanic building blocks remains underdeveloped. The scientific literature includes a limited number of reviews focused on the synthesis of fluorinated furans, with the most comprehensive reviews addressing the advances in the preparation of fluorofuran and fluoroalkylfuran derivatives covering the corresponding publications only up to 2015 [93,94]. In this paper, we discuss studies on the synthesis of fluorofuran and fluoroalkylfuran building blocks, primarily published over the last decade.
The carbon–fluorine bond formation is a challenging chemical transformation [37]. Two fundamentally distinct strategies exist for introducing fluorine-containing functional groups into furans and other heterocycles: (1) direct substitution of a hydrogen or functional group in the heterocyclic ring with fluorinated reagents or (2) cyclocondensation/cycloaddition reactions with fluorinated acyclic building blocks [18,56,66,95]. The direct introduction of fluorine and various fluoroalkyl functional groups into heteroaromatic cores can be performed with or without a catalyst through electrophilic, nucleophilic, or radical mechanisms [37,43,96,97,98,99,100]. Various environmental approaches such as the use of organocatalysts [101], electrocatalysis [40], or in-water fluorination [102] are also actively explored. The introduction of fluorinated functional groups via C–H activation eliminates the requirement for pre-functionalizing the substrate. This also enables selective functionalization at a late stage in the synthesis, which is particularly advantageous when developing new drugs and other high-value chemical products [99,103].
While approaches for the direct introduction of fluorinated functional groups into heterocycles look preferable and conceptually simpler than the use of fluorinated acyclic precursors, these methods often face challenges [18,56,57,66]. Despite the longstanding appreciation of fluorine utility, many fluorination methods still lack generality, practicality, and predictability [37,103]. Moreover, most fluorinating agents are costly, toxic, corrosive, and potentially explosive or exhibit too high reactivity, which can lead to undesired modifications in the existing functional groups [36,57,104,105,106]. Due to the electron-rich nature of the furan ring, the direct introduction of fluorinated functional groups into the furanic core predominantly occurs through electrophilic or radical substitution [93,94]. This functionalization of 2-substituted furans typically takes place selectively at the C5 position, owing to the greater stabilization of the corresponding carbocations and radicals (Scheme 2) [76,94,107,108].
2.1. Synthesis of Fluorofurans
Although methods for the synthesis of fluorofurans (compounds with the C_fur_-F bond) and fluoroalkylated furans have been developed, these methodologies often rely on non-furanic cyclic or acyclic fluorinated building blocks [93,94,109,110,111,112,113,114]. This approach has gained popularity due to its convenience and selective formation of target fluorinated molecules [57]. However, many of these methods require multi-step procedures for synthesizing specific substrates or involve costly reagents, which limits their synthetic applicability [57].
Preparation of representative fluorofurans through direct fluorination of furfural-derived substrates is summarized in Table 1. Nucleophilic fluorination via reactions with gaseous fluorine is not suitable for furan fluorination [94]. Nevertheless, some α-fluorofurans have been obtained from electron-poor furfural-derived substrates using nucleophilic fluorodenitration [78], through fluorodecarboxylation using the electrophilic fluorinating agent Selectfluor [115,116], or by employing rhodium-catalyzed heteroaryl exchange (Table 1) [117,118]. Another potential approach is the metalation-fluorination strategy, which has been effectively utilized for fluorination at both the α- and β-positions of the furan ring [119,120,121].
2.2. Synthesis of Fluoroalkylfurans
The recent methods for fluoroalkylation reactions involving furfural-derived furans can be classified into two main categories based on the mechanisms of generation and reactivity of fluoroalkyl species: (1) radical fluoroalkylation and (2) transition metal-catalyzed (cross)coupling. The mechanism of radical fluoroalkylation typically consists of three key stages (Scheme 3). The first stage involves the generation of the fluoroalkyl radical (•R_F_), which is a strong electrophile that readily reacts with electron-rich substrates such as furans I. These radicals can be produced from various fluorinated reagents, including halogenated fluoro-sources, fluorinated acids, anhydrides, sulfonyl chlorides, or hypervalent iodine reagents, typically through single electron transfer (SET) oxidation. In the subsequent stage, the •R_F_ radical inserts itself into the C5 position of the 2-substituted furanic core, resulting in the formation of a fluoroalkylated furanic radical II. Finally, the rearomatization of the furan ring leads to the desired 5-fluoroalkylated furans IV. This process generally involves a one-electron oxidation of II into the corresponding furanic cation III via an SET process, followed by deprotonation (Scheme 3). Various strategies have been employed for the radical fluoroalkylation of furans, including visible-light-mediated photocatalysis and photoredox catalysis, transition metal catalysis, and alternating current electrolysis (Table 2, Table 3, Table 4, Table 5 and Table 6).
Among the various methods for trifluoromethylation of furans, visible light-mediated radical trifluoromethylation, with or without photoredox catalysis, has recently garnered the most attention (Table 2 and Table 3) [92,122]. Photoredox-catalyzed trifluoromethylation, achieved through the generation of the electrophilic radical •CF_3_ using trifluoroacetic acid [123], trifluoroacetic anhydride (TFAA) [124,125], perfluoroarene iodine(III) trifluoroacetate [105], or trifluoromethanesulfonic anhydride [43] as CF_3_ sources has demonstrated high efficiency for the trifluoromethylation of acceptor-substituted furans such as 2-acetylfuran and 2-furoic acid derivatives (Table 2). Furfural-derived furans containing electron-donor substituents at the C2 position were trifluoromethylated through the transition-metal-mediated generation of trifluoromethyl radicals, with [38,39,106,126] or without [127,128,129] photoredox catalysis (Table 3). Zhong et al. reported a method for trifluoromethylation using perfluorocarboxylic anhydrides under metal-free conditions in the presence of urea and hydrogen peroxide [3]. This approach produced two α-trifluoromethylated furanic esters in good yields (Table 3, entries 9, 10).
Mono-, di-, and perfluoroalkyl-functionalized heterocycles are less common than aryl-fluorinated (Ar-F) or aryl-trifluoromethylated (Ar-CF_3_) compounds, but they remain of interest due to their biological activity and applications in materials science [134,135]. Representative and efficient methods for fluoroalkylation of both electron-poor (Table 4 and Table 5) and electron-rich (Table 6) furans are outlined below. The nucleophilic fluorinating reagent Deoxo-Fluor exhibited significant selectivity in the deoxyfluorination of 5-nitro-2-furaldehyde to produce 2-(difluoromethyl)-5-nitrofuran (Table 5, entry 8), as well as in the fluorination of several 2,5-disubstituted furanic aldehydes [136]. Additionally, Zhang et al. utilized another nucleophilic fluorinating reagent DAST for the direct difluoromethylation of furfuryl acetate at the C5 position (Table 6, entry 13) [137].
Transition metal-mediated C-H functionalization and radical visible light-mediated fluoroalkylation, with or without photoredox catalysis, demonstrated high efficiency for the introduction of fluoroalkyl groups into furans. Using these methods, mono-, di-, and perfluoroalkylated furanic building blocks were obtained with notable efficiency from electron-poor furans including 2-furoates (Table 4), furfural, 2-furoic acid, and other derivatives (Table 5) [14,140,150,151] and electron-rich furans such as 2-methylfuran, furfural-based dioxolane acetal, furfuryl alcohol and derivatives (Table 6). Coupling strategies involving furyl iodides [146,152,153] or sulfonyl chlorides [149] were also effectively employed for the fluorination of acceptor-substituted furans (Table 4, entries 9 and 12, and Table 5, entries 4–7).
As shown in Table 2, Table 3, Table 4, Table 5 and Table 6, most fluoroalkylation methods were tested on either C2 donor- or acceptor-substituted furanic substrates. Some methodologies, such as Pd-catalyzed difluoromethylation using ethyl 2-bromo-2,2-difluoroacetate [140], Cu-catalyzed perfluoroalkylation with IC_4_F_9_ [142], and electrocatalytic trifluoromethylation [131] demonstrated considerable efficiency for both donor- and acceptor-substituted furans. However, the applicability of many reported fluoroalkylation systems of furans is not fully established, as the range of tested furanic substrates was often limited to highly stable or non-functionalized compounds like 2-methylfuran [14,36,106,126,127,129,134,161,162], methyl 2-furoate [146,148], or 2-acetylfuran [151]. In some cases, fluoroalkylation resulted in significantly lower yields compared to other heteroaromatic substrates, or even failed [22,163,164,165].
Synthesis of β-fluorinated and β-fluoroalkylated furans through direct fluoroalkylation presents challenges; however, it can be achieved using fluorinated acyclic precursors [70,169,170,171,172,173]. On the other hand, the dienic nature of the furan ring offers the construction of fluorofurans via Diels–Alder chemistry. Regioselective hydrogenation of the unsubstituted double bond in Diels–Alder adducts 7 and 8 formed from electron-rich furans and hexafluorobutyne, followed by temperature-induced elimination of ethylene, resulted in β,β-bis-trifluoromethylated furans 9 and 10 in good overall yields (Table 7) [174].
In the same study, bis-trifluoromethylated furans 11–15 containing electron-accepting functional groups at the C2 position were synthesized in a single step through high-temperature reactions with heptafluorobutene (Table 7). This alkene served as a strong dienophile reacting with 2-furonitrile, 2-furoic acid or its esters, and even with furfural, producing the desired fluorinated trisubstituted furans in good yields [174].
3. Application of Furfural-Derived Fluorofurans as Building Blocks in Drug Development
Introduction of fluorinated furan derivatives into biologically active scaffolds has garnered significant attention [62,63]. The polyfunctional nature of furfural-derived furans, which allow several reactivity patterns such as the modification of the substituents at the C2 position, the functionalization of the C5-H bond, and the dienic reactivity of the furan ring, provides efficient and diverse chemical access to various fluorofuran or fluoroalkylfuran derivatives. A possible reaction map for the synthesis of fluorinated furanic building blocks starting from furfural is presented in Scheme 4. The introduction of fluorinated furan moieties into biologically active scaffolds can be achieved through reductive amination with fluorinated furanic aldehydes (pathway A). The 2-(bromomethyl)-5-(fluoroalkyl)furans, obtainable through the bromination of corresponding 2-(methyl)-5-(fluoroalkyl)furans, can serve as an alkylating agent or substrate in various coupling reactions (pathway B) [175,176,177,178,179]. C5-fluorinated furoic acids can also act as convenient building blocks for the introduction of fluorofuran or fluoroalkylfuran groups via acylation (pathway C). Another potential approach involves the use of halogenated or sulfonated α-fluorofurans in cross-coupling reactions (pathway D). Functionalized furans containing fluorinated functional groups at the β-positions may be potentially synthesized using the Diels–Alder strategy (pathway E). Recent and representative examples of the application of fluorofuran and fluoroalkylfuran reagents in drug development using these approaches are discussed below.
α-Fluorofuran 16 with a boronic acid pinacol ester group can be synthesized from 5-bromo-2-furoic acid through decarbonylative fluorination, followed by a reaction with pinacol diborate [180]. Cross-coupling of 16 was employed to introduce the 2-fluorofuran fragment into targeted molecules in the development of inhibitors of MCL1 for cancer therapy (Scheme 5) [65,181]. Compound 2 (Table 1) binds to the BH3-binding groove of MCL1 with high selectively and affinity [65]. In later studies, a number of similar 4-fluorofur-2-yl (17) and 4-fluorophenyl (18) derivatives were discovered. Despite the lower activity of 4-fluorophenyl derivatives 18 in vitro, a fluorophenyl analogue demonstrated the in vivoefficacy comparable to that of fluorofuryl derivatives 17, prompting further research on fluorophenyl derivatives 18 [181].
Pinacol boronic ester 16 was also utilized in the synthesis of compound 19a, which is a α-fluorofuran analogue of manogepix (APX001A), the active moiety of a novel experimental antifungal drug fosmanogepix (Scheme 6). The antifungal activity of 19a was evaluated against Cryptococcus neoformans and Cryptococcus gattii [182]. Compound 19a exhibited notably lower MIC values and a longer half-life compared to manogepix and their furanic nonfluorinated analogue 19b. Therefore, it can be concluded that the incorporation of a fluorine atom into the furan fragment of the manogepix analogue 19b enhances its metabolic stability.
Various 1,2,3,4-tetrahydroisoquinoline-3-carboxylic acids, including 3-(5-fluorofuryl)-substituted derivatives 21(a,b), were synthesized and evaluated for their ability to partially activate peroxisome proliferator-activated receptor gamma (PPARγ), which plays a key role in lowering the blood sugar level in patients with type 2 diabetes [183]. In this series, compound 21b containing an indanyl group exhibited reduced affinity for the PPARγ receptor in comparison to its non-fluorinated counterpart that was selected as the lead compound for further studies. The compounds 21(a,b) were synthesized from 3-(5-fluorofuryl)acrylic acid 20, which was obtained from ethyl 5-bromofuran-2-carboxylate through decarbonylative fluorination using Selectfluor (Scheme 7) [183].
5-Fluorofuranic amides 22 and 23 were synthesized with the aim of searching for new effective selective agonists of α4β2 nicotinic acetylcholine receptors (nAChR) [184]. These amides were obtained through the acylation of corresponding Boc-protected amines with 5-fluoro-2-furoic acid using HBTU as a carboxylic group activator, followed by Boc deprotection (Scheme 8) [184]. SAR studies indicated that furoamide 22 exhibited sufficiently high efficacy as an α4β2 nAChR-selective agonist among other studied heterocyclic amides. However, the significant undesirable activation of nAChR receptors specifically located in the ganglia hindered the potential of 5-fluoro-2-furyl derivatives 22 and 23 as lead compounds for further in vivotesting.
To mitigate potential genotoxicity associated with the formation of nitrofuran metabolites, the nitro group in furanic orally available UT inhibitor 24 was replaced with more stable fluorine or difluoromethyl group [185]. These new compounds 25(a,b) were synthesized by condensing an N-(4-aminophenyl)acetamide with fluorinated furan-2-carboxylic acids in the presence of a base and HATU (Scheme 9). Unfortunately, the substitution of the nitro group with fluorine or difluoromethyl group resulted in a significant reduction in biological activity for this class of compounds.
The α-trifluoromethylfuran derivatives 27(a–d) displayed significantly higher antifungal activity against C. neoformans compared to their non-fluorinated furanic analogs. However, these compounds were less potent than their thiophene counterparts. Although in subsequent tests on fungicidal or fungistatic activity, as well as on selectivity against fungi/human cells, none of trifluoromethylated furans were sufficiently active and selective to be included in the leader compound set, the results of these experiments indicate the importance of substituents in furan moieties for high antifungal activity [186]. The abovementioned derivatives 27(a–d) were obtained from the corresponding 5-trifluoromethylfurane hydrazide 26, which was synthesized starting from 2-trifluoromethylfuran-5-carboxylic acid (Scheme 10).
Using a series of 5-(5-furan-2-yl-pyrazol-1-yl)-1H-benzimidazole derivatives that inhibited the assembly of the HIV-1 capsid, the effect of site-selective modifications at N1, C2 and C16 of this scaffold was clarified. Replacing the methyl substituent at C16 with a trifluoromethyl group (compound 33) produced a six-fold gain in the antiviral potency significantly affecting the IC_50_ values [187]. The key stage of the synthesis of these 5-(5-furan-2-ylpyrazol-1-yl)-1H-benzimidazoles was the acidic condensation of hydrazine 31 with fluorinated furanic diketone 30, which in turn was obtained from 5-(trifluoromethyl)furan derivative 28 (Scheme 11).
Phthalazin-1(2H)-ones with furan as a side substituent showed the highest levels of antiproliferative activities against olaparib- and talazoparib-resistant Capan-1 cells [61]. In attempts to find the optimal substituents at the α-carbon of the furan ring, derivatives 38(a-e) containing the furan substituted at the α-carbon position by various fluoroalkyl moieties were obtained (Scheme 12).
The initial building block for the synthesis of 38a was α-trifluoromethylated furanic ester 34. This compound was converted into triazole 35 through a two-step process involving modification to hydrazide 26, followed by cyclization. The α-difluoromethyl fragments in 38b and 38(c-e) were introduced via fluorination at α-position of the furanic ring in intermediates 36 and 37, respectively. Among these compounds, water-soluble derivatives 38(c-e) demonstrated improved inhibitory activity, but reduced potency towards drug-resistant cells (olaparib- and talazoparib-resistant Capan-1 cells) [61]. Thus, unfortunately, none of these fluorinated furan structures emerged as the leader compound in this study.
4. Conclusions
Although numerous methods have been reported for introducing fluorine or fluorine-containing functional groups into various aromatic and heteroaromatic compounds, direct functionalization approaches for furfural-derived furans remain underdeveloped. Current methodologies for synthesizing α-fluorofurans and α-fluoroalkylfurans still suffer from low yields, limited generality, or reliance on an expensive reagent. Furthermore, furans containing fluorinated functional groups at the β-positions were synthesized from furfural-derived substrates only through indirect methods. The existing fluoroalkylation techniques were typically tested on a limited range of relatively stable furfural-derived furan substrates, such as 2-methylfuran, methyl 2-furoate, and 2-acetylfuran.
Fluorinated and fluoroalkylated furans, benefiting from the unique properties of both furan and fluorine functionalities, hold significant promise for drug design. However, none of these compounds have yet been approved for clinical use by the Food and Drug Administration (FDA). On the other hand, fluorination significantly increases the exothermic effect of Diels–Alder reactions and lowers the activation energy barriers, which is more pronounced at the α-position of furan compared to the β-position [188,189]. Consequently, incorporating fluorofurans or fluoroalkylfurans into the structures of Diels–Alder-based dynamers could substantially influence their thermal and chemical stability. However, our literature review revealed no reported examples of fluorinated furans being applied in the development of Diels–Alder dynamers.
To achieve more sustainable fluorinated polymers and pharmaceuticals, it is essential to develop more versatile and efficient methods for introducing fluorinated functional groups into furan derivatives. Key considerations include the applicability of these methods to a range of furfural-derived donor- or acceptor-substituted furan substrates, including those containing sensitive functional groups. Establishing such methodologies would not only expand synthetic possibilities but also promote the active and efficient utilization of plant biomass in drug design and materials science through the development of fluoro-functionalized furans.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Budisa N. Kubyshkin V. Schulze-Makuch D. Fluorine-rich planetary environments as possible habitats for life Life 2014437438510.3390/life 403037425370378 PMC 4206852 · doi ↗ · pubmed ↗
- 2Ismail F.M.D. Important fluorinated drugs in experimental and clinical use J. Fluorine Chem.2002118273310.1016/S 0022-1139(02)00201-4 · doi ↗
- 3Zhong S. Hafner A. Hussal C. Nieger M. Bräse S. Metal-free radical perfluoroalkylation of (hetero)arenes RSC Adv.201556255625810.1039/C 4RA 13430 C · doi ↗
- 4Rizzo C. Amata S. Pibiri I. Pace A. Buscemi S. Palumbo Piccionello A. FDA-Approved Fluorinated Heterocyclic Drugs from 2016 to 2022 Int. J. Mol. Sci.202324772810.3390/ijms 2409772837175436 PMC 10178595 · doi ↗ · pubmed ↗
- 5Han J. Kiss L. Mei H. Remete A.M. Ponikvar-Svet M. Sedgwick D.M. Roman R. Fustero S. Moriwaki H. Soloshonok V.A. Chemical Aspects of Human and Environmental Overload with Fluorine Chem. Rev.20211214678474210.1021/acs.chemrev.0c 0126333723999 PMC 8945431 · doi ↗ · pubmed ↗
- 6Gillis E.P. Eastman K.J. Hill M.D. Donnelly D.J. Meanwell N.A. Applications of Fluorine in Medicinal Chemistry J. Med. Chem.2015588315835910.1021/acs.jmedchem.5b 0025826200936 · doi ↗ · pubmed ↗
- 7Shah P. Westwell A.D. The role of fluorine in medicinal chemistry J. Enzyme Inhib. Med. Chem.20072252754010.1080/1475636070142501418035820 · doi ↗ · pubmed ↗
- 8Bohm H.J. Banner D. Bendels S. Kansy M. Kuhn B. Muller K. Obst-Sander U. Stahl M. Fluorine in medicinal chemistry Chem Bio Chem 2004563764310.1002/cbic.20030102315122635 · doi ↗ · pubmed ↗