Tissue-specific mitochondrial DNA, MT-TF, pathogenic variants in mitochondrial myopathies
Sylvia Rose, Aurélien Trimouille, Didier Lacombe, Edoardo Malfatti, Zahra Assouline, Julie Steffann, Isabelle Desguerre, Arnold Munnich, Agnès Rötig, Giulia Barcia

TL;DR
This paper highlights the importance of analyzing mitochondrial DNA in muscle tissue rather than blood for diagnosing mitochondrial myopathies due to variable genetic mutations across tissues.
Contribution
The study demonstrates that mtDNA variants in muscle tissue can be missed when only blood samples are analyzed.
Findings
MT-TF variants were found in muscle tissue with high heteroplasmy levels, even when blood samples showed no mutations.
Clinical symptoms varied among patients, emphasizing the need for tissue-specific genetic testing.
Muscle DNA analysis is crucial for accurate diagnosis when blood tests are negative but clinical suspicion remains high.
Abstract
Mitochondrial myopathies are progressive muscle disorders caused by impaired mitochondrial oxidative phosphorylation, leading to reduced adenosine triphosphate production. Skeletal muscles have a high energy demand and are often the first to be affected. In addition to muscular symptoms (muscle weakness, effort intolerance, fatigue), the disease can affect the central and peripheral nervous systems, as well as the heart, liver, kidneys and endocrine system (diabetes). Molecular genetic diagnostic is currently based on leukocyte DNA obtained from blood samples, considered less invasive than muscle biopsy. We report four patients from three families with mitochondrial myopathy associated with ptosis, sensorineural hearing loss, epilepsy, tubulointerstitial nephropathy and cardiomyopathy. Genetic studies identified MT-TF variants (m.586G > A, m.601G > A, m.616 T > C) with highly variable…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
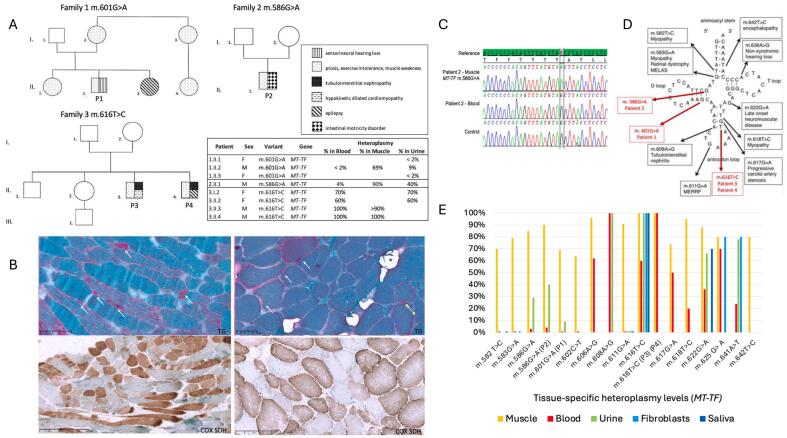 Figure 1
Figure 1Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsMitochondrial Function and Pathology · Metabolism and Genetic Disorders · Genetic Neurodegenerative Diseases
Introduction
1
Mitochondrial myopathies (MM) are progressive muscle disorders with a prevalence of 1/4300 [1], caused by impaired mitochondrial oxidative phosphorylation (OXPHOS), leading to reduced adenosine triphosphate (ATP) production. Skeletal muscles have a high energy demand and are often the first to be affected [2]. In addition to muscular symptoms (muscle weakness, effort intolerance, fatigue), the disease can affect the central and peripheral nervous systems, as well as the heart, liver, kidneys and endocrine system.
MM can be caused by pathogenic variant affecting either mitochondrial or nuclear DNA [3]. The clinical expression of mitochondrial DNA (mtDNA) mutations depends on the percentage of wild type and mutant mtDNA coexisting in any given cell, known as heteroplasmy. Mitochondrial transfer RNA (tRNA) gene mutations are associated with a wide range of pathologies, from isolated myopathy, to multisystem disorders, including encephalopathy, epilepsy, gastrointestinal (GI) dysmotility, hearing loss and life-threatening cardiomyopathy [4]. The diagnostic of MM traditionally relies on specific histological markers in muscle biopsy such as ragged red fibers (RRF) or cytochrome c oxidase (COX) negative/succinate dehydrogenase (SDH) positive fibers and measurement of OXPHOS enzymatic activity in muscle. Nowadays, definitive diagnostic is usually based on mtDNA sequencing performed on blood leukocyte DNA [5].
We report three families with MM due to three different pathogenic variants in the mitochondrial MT-TF gene, which encodes mitochondrial tRNA^Phe^. In MM, heteroplasmy level can vary significantly in different tissues of a same patient. Clinicians should be aware of this variability to avoid missing a molecular diagnosis of MM.
Methods
2
Subjects
2.1
The four patients were recruited in two genetic departments (Paris and Bordeaux) belonging to the CARAMMEL French reference network for mitochondrial diseases.
Clinical data including family history, psychomotor development, neurological examination, biochemical and metabolic work-up, and neuroimaging studies were obtained for all patients.
Patient 2 was previously reported [6].
Muscle histopathology
2.2
Muscle biopsies from patients 1–3 were frozen in liquid nitrogen. Cryostat sections were used for hematoxylin-eosin-safran (HES), Gömöri trichrome and double COX-SDH enzymatic staining. Myosin heavy chain expression was assessed using monoclonal antibodies against fast and slow isoforms (myosin fast/slow).
OXPHOS spectrophotometric analysis in muscle and heart
2.3
OXPHOS spectrophotometric analysis was performed as previously described [7].
Genetic studies
2.4
Total DNA was extracted from peripheral blood, urinary epithelial cells, skeletal muscle cells and saliva using Qiagen or Autopure DNA purification kit and sequenced using next-generation sequencing. Single nucleotide variants (SNVs) were confirmed by Sanger sequencing and segregation analysis was completed in each family (Fig. 1C).Fig. 1. Patients, phenotypes, genetic variants in MT-TF gene, mutant loads and segregation of the MT-TF variants.A. Families' pedigrees. B. Histological features of muscle from patient 1 (left) and patient 2 (right). Upper panel: Gömöri trichrome staining revealed a typical aspect of mitochondrial myopathy with numerous RRF (white arrows) with large lipid droplets in most fibers (*). Lower panel: Sequential COX-SDH reaction revealed a mosaic pattern of COX-deficiency. C. Sanger sequencing of MT-TF showing the m.586G > A substitution in muscle DNA but not in blood leukocytes of P2. D. Schematic diagram of the mitochondrial tRNA^Phe^ cloverleaf structure with the variations reported in this article (in red) and previously reported mutations. Adapted from [10] E. Tissue-specific heteroplasmy levels for each MT-TF point mutation reported in the literature.Fig. 1
mtDNA was amplified in two fragments by long range PCR (8.2 Kb and 8.3 Kb). Libraries were prepared from 1.5 μg of PCR sheared with a Covaris S2 Ultrasonicator. DNA was sequenced on Illumina MiSeq (Illumina Inc., San Diego, CA) generating 2 × 130 paired-end reads aligned against human mtDNA (NC_ 012920.1) using BWA Burrows-Wheeler Aligner.
Ethics statement
2.5
This study adhered to the Declaration of Helsinki and was approved by our institutional review board. Informed consent for diagnostic and research studies was obtained for each patient.
Results
3
Case reports
3.1
Family 1
3.1.1
Patient 1 (male) was 66 years old at the time of diagnosis. He presented bilateral ptosis, exercise intolerance and sensorineural hearing loss. Metabolic work-up revealed a normal plasma lactate and CPK levels. He had an older sister presenting with ptosis and exercise intolerance and a younger sister with infantile onset epilepsy and muscle fatigability. Their mother, maternal aunt and cousin had ptosis and effort intolerance (Fig. 1A).
Family 2
3.1.2
Patient 2 (male) was 14 years old at the time of diagnosis [6]. He is the only child of unrelated parents with unremarkable family history (Fig. 1A). He was born after a full-term pregnancy, a normal delivery (birth weight 2535 g; height 49.5 cm; head circumference 34 cm) and had normal psychomotor development. At the age of 8, he started to present diffuse myalgias, nocturnal cramps, exercise intolerance after short periods (<5 min), abdominal pain and vomiting following physical exertion. At the last clinical examination at the age of 17, he weighted 34 Kg with a height of 1.67 m (BMI 12.2 kg/m^2^), has reduced muscle mass, diminished strength in the lower limbs (2/5), trunk and limb-girdle weakness, difficulty sitting unaided, and progressive scoliosis. The score obtained in the 6-min walk test was 344 m (normal: 0–900 m), with a high fatigue index of 18 %. Cerebral MRI and cardiac ultrasound were normal. Metabolic workup revealed a high plasma lactate level of 4.8 to 6.9 mmol/L (normal: 0.5 to 1.5 mmol/L) and a high lactate/pyruvate ratio (L/P = 61; normal: <20), normal CPK and plasma amino acid chromatography. EMG and motor/sensory nerve conduction were normal.
Family 3
3.1.3
Patient 3 (male) was 28 years old at the time of diagnosis. Family history is not informative, and his parents are not related (Fig. 1A). He was born at term of a regular pregnancy with normal measurements (birth weight 3530 g; height 50 cm; head circumference 35 cm) and had a normal psychomotor development. He presented iron-deficient anemia in infancy.
At age of 4, he presented with pallor, profound asthenia and anorexia, which led to the diagnosis of severe cardiac insufficiency due to dilated cardiomyopathy, hypertension (180/120 mmHg), anemia (8.8 g/dL), renal insufficiency (elevated creatine level 310 μmol/L and urea 38 mmol/L) without proteinuria. He also presented fatigability, exercise intolerance, muscle weakness, and ptosis. Cerebral MRI was normal. A metabolic workup revealed elevated plasma lactate (3.3 mmol/L, normal: 0.5 to 1.5 mmol/L), elevated CSF lactate (2.15 mmol/L; normal: < 2 mmol/L), normal CPK and normal plasma amino acid chromatography. Hypokinetic dilated cardiomyopathy progressed requiring heart transplantation at the age of 15. Tubulointerstitial nephropathy worsened requiring renal transplant at the same age.
Patient 4 (male) is the younger brother of patient 3, aged 25 at the time of diagnosis. He presented tubulointerstitial nephropathy, progressing to hemodialysis-treated end-stage renal failure. He was treated by Lamictal and Rivotril for generalized epileptic seizures following dialysis sessions. Cardiac features include ventricular bigeminy with no other rhythm or conduction disorders, and no cardiac function abnormalities. Neurologically, he complains of fatigability with effort intolerance and muscle weakness associated with ptosis.
Muscle histopathology
3.2
Muscle biopsy study of P1 and P2 (Fig. 1B) reveals a typical appearance of mitochondrial myopathy, with numerous ragged red fibers (RRF) shredded by Gömöri trichrome and COX-negative and SDH-positive fibers. P2 biopsy also showed lipid droplets. P3 muscle biopsy did not show significant abnormality apart the low intensity of the COX reaction (not shown). P4 did not have a muscle biopsy.
OXPHOS enzyme activities
3.3
Investigation of respiratory chain complex activities showed combined deficiency of complexes I, III and IV in muscle from P2. Complex II and citrate synthase were increased suggesting a compensatory upregulation. An isolated Complex IV defect was detected in muscle and heart of P3.
Molecular analysis of mtDNA
3.4
NGS sequencing revealed the presence of a heteroplasmic m.601G > A variant in MT-TF, with high mutation load in skeletal muscle of P1 (69 %) while barely detectable in urinary epithelia (9 %) and leukocytes (<2 %). This variant has never been reported so far, is absent from Mitomaster and mtDB databases, and modifies an evolutionarily highly conserved nucleotide. Thus, m.601G > A variant was classified as likely pathogenic. The mutation was <2 % in blood DNA of his two sisters. In P2, a heteroplasmic m.586G > A variation in MT-TF was detected in skeletal muscle (>90 % mutant load) and urinary epithelial cells (40 %) while barely detectable mutations were found in circulating leukocytes (4 %). This variation could not be detected in circulating leukocytes and urinary epithelial cells of the mother. This variation affects a phylogenetically conserved nucleotide, located in the D-stem and is listed in the Mitomap database as likely pathogenic. [8]
P3 and P4 presented a m.616 T > C variant in MT-TF, that was heteroplasmic (>90 % mutant load) and homoplasmic in blood leukocytes of P3 and P4 respectively. This variant is reported in Mitomap database as likely pathogenic, modifying an evolutionarily highly conserved nucleotide. In asymptomatic mother and sister, this variant was detected in blood (60–70 %) and urine (60–70 %).
Discussion
4
We report a novel likely pathogenic MT-TF (m.601G > A) variant in a patient presenting MM with exercise intolerance, muscle weakness, fatigability, ptosis, and sensorineural hearing loss. Moreover, we also describe 3 patients carrying two rare MT-TF pathogenic variants, m.586G > A and m.616 T > C, only reported in a few patients (Table 1). Pathogenic mutations of tRNAs have been shown to affect global tRNA structure, tRNA processing, modification, aminoacylation, and translation efficiency [9]. The m.586G > A and m.601G > A variants, located in the D branch, could affect tRNA stability, as previously demonstrated for the m.582 T > C and m.583G > A variants [9,10]. The m.616 T > C variant modifies an evolutionarily conserved nucleotide, the only pseudo-uridine in mitochondrial tRNA^Phe^, and disrupts the last nucleotide pairing before the anticodon loop (Fig. 1D).Table 1. Overview of clinical features of individuals carrying variants in MT-TF and tissue-specific mutant load reported in MitoMap.Table 1. VariantReferencePhenotypeAge at onset (yrs)Age at diagnosis (yrs)PathogenicityMutant loadHeredityMuscleBloodUrineFibroblastsSalivam.582 T > CMoslemi et al. 2004Mitochondrial myopathy, ptosis5070Possiblypathogenic70 %0 %NDND0 %de novom.583G > AHanna et al.1998MELAS123258 %0 %ND0 %NDde novom.583G > ADarin et al. 2006Mitochondrial myopathy, retinopathy1417Pathogenic79 %0 %ND0 %NDde novom.586G > AYoung et al.2010Mitochondrial myopathy, encephalopathy, lactic acidosis, gastrointestinal dysmotility,Tubulointerstitial nephropathy4557Likelypathogenic85 %3 %29 %NDNDmaternalm.586G > AD'Aco et al. 2013Encephalopathy, regression, lactic acidosis, gastrointestinal dysmotility, Tubulointerstitial nephropathy1heteroplasmicheteroplasmicNDNDNDde novom.586G > ABarcia et al.2019Patient 2814Likelypathogenic>90 %4 %40 %NDNDde novom.591C > TViering et al. 2022Tubulointerstitial nephropathy, muscle weakness, epilepsyLikelypathogenicND98 %NDNDNDm.601G > AThis reportPatient 166Likelypathogenic69 %0 %9 %NDNDm.602C > TSakiyama et al.2011Mitochondrial myopathy, ptosis, dysarthria, cataract, SNHL6373Likelypathogenic64 %0 %NDNDNDmaternalm.606 A > GKleinle et al. 1998Rhabdomyolysis, myoglobinuria153096 %62 %NDNDNDmaternalm.608 A > GTzen et al.2001MELAS, epilepsyTubulointerstitial nephropathy01LikelypathogenicND100 %100 %NDNDmaternalm.610 T > CIndelicato et al. 2023Myoclonic epilepsyDevelopmental regressionNORSE729NDND97 %97 %NDND100 %NDNDNDmaternalm.611G > AMancuso et al. 2004MERRF2042Likelypathogenic91 %0 %0 %0 %0 %de novom.616 T > CViering et al.2022Tubulointerstitial nephropathy, muscle weakness, epilepsyND100 %NDNDNDNDm.616 T > CConnor et al.,2017Tubulointerstitial nephropathy muscle weakness, epilepsyND100 %NDNDNDNDm.616 T > CZsurka et al. 2010Epileptic encephalopathy, Tubulointerstitial nephropathy117Pathogenic100 %100 %100 %99 %NDmaternalm.616 T > CLorenz et al. 2020.Epileptic encephalopathy, Tubulointerstitial nephropathy35NDNDNDNDNDNDm.616 T > CXu et al.2022Tubulointerstitial nephropathy, muscle weakness, epilepsyND>90 %100 %100 %100 %NDm.616 T > CThis reportPatient 3428PathogenicND>90 %NDNDNDmaternalm.616 T > CThis reportPatient 425PathogenicND100 %NDNDNDmaternalm.616 T > GZsurka et al.2010Myoclonic epilepsy, Tubulointerstitial nephropathy15Likelypathogenic100 %100 %NDNDNDNDm.617G > AIizuka et al.2009MELAS, carotid artery stenosis840Likelypathogenic74 %50 %NDNDNDmaternalm.618 T > CKleinle et al. 1998Mitochondrial myopathy2636Possiblypathogenic95 %20 %NDNDNDmaternalm.618 T > GYarham et al.2011Mitochondrial myopathy, PEO, ptosis, dysphagia25Likelypathogenic76 %0 %0 %NDNDde novom.622G > ADeschauer et al. 2006Exercice intolerance, SNHL626688 %36 %66 %ND70 %maternalm.625 G > ASudo et al.2011Epilepsy, SNHL611Likelypathogenic80 %70 %ND80 %NDNDm.628C > TDowlati et al.2013SNHL27NDNDNDNDNDmaternalm.636 A > GKonings et al.2008SNHL1NDNDNDNDNDNDm.641 A > TItkis et al.2019Myoclonic epilepsy, motor regression with spastic tetraplegia8PossiblypathogenicND24 %78 %80 %NDNDm.642 T > CValente et al. 2009Mitochondrial myopathy, encephalopathy, PEO, ptosis, SNHL, peripheral neuropathy, gait ataxia47Possiblypathogenic80 %NDNDNDNDNDm.643 A > GViering et al.2022Tubulointerstitial nephropathy, muscle weakness, epilepsyVUSND100 %NDNDNDNDList of abbreviations. MELAS: Mitochondrial Encephalomyopathy, Lactic Acidosis and Stroke-like episodes; MERRF: Myoclonic Epilepsy with Ragged Red Fibers; ND: not determined; NORSE: New-Onset Refractory Myoclonic Status Epilepticus; PEO: Progressive External Ophthalmoplegia; SNHL: Sensorineural Hearing Loss; VUS: Variants of Unknown Significance.
Patients with mitochondrial tRNA mutations have complex clinical symptoms with highly heterogeneous phenotype. MT-TF pathogenic variants have been associated to a variety of clinical symptoms, including myopathy, ptosis, sensorineural hearing loss, epilepsy and tubulointerstitial nephropathy. The age of onset of symptoms and their severity is variable, as is often the case with mitochondrial diseases (Table 1).
A previous report described a 16-month-old girl with the MT-TF m.586G > A variant (heteroplasmic in muscle and blood) associated with failure-to-thrive, developmental regression, persistent lactic acidosis, hypotonia, GI dysmotility and renal impairment with chronic tubulointerstitial fibrosis, progressing to end-stage renal disease [11]. This MT-TF m.586G > A variant has also been reported in a 57 yrs-old woman with progressive neurodegenerative disorder, akinesia-rigidity, abnormal movements, dementia, and psychiatric disorder [12]. This patient also had lactic acidosis, GI dysmotility, renal failure. No MM was observed but muscle biopsy showed markers of MM. Heteroplasmy level was 3 % in blood and 85 % in muscle, comparable with P2 heteroplasmy level for this specific m.586G > A variant.
The m.616 T > C variant has been described in patients presenting epilepsy and tubulointerstitial kidney disease. Muscle weakness was often reported without further investigation. Analysis of muscle histology when performed showed decreased overall COX staining [13].
Our report aims to emphasize that MM associated with mitochondrial tRNA^Phe^ mutations are not uncommon and are probably underdiagnosed because of the highly variable mutant load in different tissues. Indeed, genetic analyses are most often performed on leukocyte DNA as muscle biopsy remains an invasive procedure. High-throughput sequencing of mtDNA, with a coverage of >1000 reads of each nucleotide, can detect a low load of mutant mtDNA allowing genetic diagnosis. However, in P1 and P2, the mutation was not detectable in leukocytes, which has also been reported for other MT-TF mutations with a mutant load less than 2 % [4,6,9,10,[13], [14], [15], [16], [17], [18], [19]]. Similarly, certain variants of the MT-TF gene have been investigated in several tissues to assess the variability of heteroplasmy levels (Fig. 1E). Therefore, a strong clinical suspicion of MM should prompt to test additional tissues, especially muscle, even if no mtDNA variant could be detected in leukocyte. Samples of urine or saliva are readily available to assess heteroplasmic mtDNA variations and are easier to collect than muscle or fibroblasts. Thus, the presence of 40 % mutated molecules in the urine of P2 reinforces the diagnostic hypothesis.
It seems essential to continue the precise clinical and histological study of mitochondrial tRNA variants, in conjunction with existing molecular databases to confirm diagnosis.
CRediT authorship contribution statement
Sylvia Rose: Conceptualization, Writing – original draft, Writing – review & editing. Aurélien Trimouille: Conceptualization, Supervision, Validation. Didier Lacombe: Conceptualization, Supervision, Validation. Edoardo Malfatti: Supervision, Validation. Zahra Assouline: Supervision, Validation. Julie Steffann: Supervision, Validation. Isabelle Desguerre: Supervision, Validation. Arnold Munnich: Supervision, Validation. Agnès Rötig: Conceptualization, Supervision, Validation, Writing – review & editing. Giulia Barcia: Conceptualization, Supervision, Validation, Writing – review & editing.
Declaration of competing interest
None.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Myopathie mitochondriale | AFM Téléthon [Internet][cited 2023 Aug 7]. Available from:https://www.afm-telethon.fr/fr/fiches-maladies/myopathie-mitochondriale 2023
- 2Larsson N.G.Oldfors A.Mitochondrial myopathies Acta Physiol. Scand.171320013853931141215210.1046/j.1365-201x.2001.00842.x · doi ↗ · pubmed ↗
- 3Tuppen H.A.L.Blakely E.L.Turnbull D.M.Taylor R.W.Mitochondrial DNA mutations and human disease Biochim. Biophys. Acta 179722010 Feb 1131281976175210.1016/j.bbabio.2009.09.005 · doi ↗ · pubmed ↗
- 4Yarham J.W.Elson J.L.Blakely E.L.Mc Farland R.Taylor R.W.Mitochondrial t RNA mutations and disease Wiley Interdiscip. Rev. RNA 1220103043242193589210.1002/wrna.27 · doi ↗ · pubmed ↗
- 5Meola G.Bugiardini E.Cardani R.Muscle biopsy J. Neurol.25942012 Apr 6016102180525610.1007/s 00415-011-6193-8 · doi ↗ · pubmed ↗
- 6Barcia G.Assouline Z.Pennisi A.Steffann J.Boddaert N.Gitiaux C.Expanding the clinical spectrum of MTTF mutations Mol. Genet. Metab. Rep.212019 Dec 110050110.1016/j.ymgmr.2019.100501 PMC 670667731463198 · doi ↗ · pubmed ↗
- 7Barcia G.Khirani S.Amaddeo A.Assouline Z.Pennisi A.Boddaert N.Evidence of diaphragmatic dysfunction with severe alveolar hypoventilation syndrome in mitochondrial respiratory chain deficiency Neuromuscul. Disord. NMD 3072020 Jul 5935983265495210.1016/j.nmd.2020.06.002 · doi ↗ · pubmed ↗
- 8Web Home < MITOMAP < Foswiki [Internet][cited 2023 Oct 23]. Available from:https://www.mitomap.org/MITOMAP