Benchmarking Nanopore Sequencing for CLN2 (TPP1) Mutation Detection: Integrating Rapid Genomics and Orthogonal Validation for Precision Diagnostics
Betül Teker, Gökce Akan, Hasan Hüseyin Kazan, Özge Özgen, Suzin Tatonyan, Mehmet Cihan Balci, Meryem Karaca, Fulya Kurekci, Edibe Pembegül Yıldız, Olcay Güngor, Adnan Deniz, Asuman Gedikbasi, Fatmahan Atalar, Gülden Fatma Gokcay, Mehves Poda

TL;DR
This study evaluates the use of nanopore sequencing for detecting mutations in the TPP1 gene, which causes CLN2 disease, and confirms its effectiveness in a Turkish patient cohort.
Contribution
The study is the first to report specific variant configurations in CLN2 disease and validates nanopore sequencing for this purpose in a high-consanguinity population.
Findings
Four pathogenic TPP1 variants were identified, including novel homozygous and compound heterozygous configurations.
ONT-LRS showed high concordance with Sanger sequencing and correlated with reduced TPP1 enzyme activity in affected individuals.
Regional variation in TPP1 mutation patterns was observed, with the c.622C>T variant being the most prevalent in the Turkish cohort.
Abstract
CLN2 disease (neuronal ceroid lipofuscinosis type 2) is an ultra-rare lysosomal storage disorder caused by mutations in the TPP1/CLN2 gene, resulting in impaired tripeptidyl peptidase 1 (TPP1) activity. The timely initiation of enzyme replacement therapy is pivotal for attenuating progressive and irreversible neurodegeneration. This study aimed to benchmark the performance of Oxford Nanopore long-read sequencing (ONT-LRS) for targeted TPP1 mutation detection in a Turkish CLN2 cohort and to assess its concordance with orthogonal validation methods, including Sanger sequencing and enzymatic activity assays. Using a custom-designed primer panel, the entire TPP1 gene (6846 bp) was sequenced on the Oxford Nanopore (ONT) MinIon platform in seven clinically confirmed CLN2 index patients and sixteen unaffected family members. Detected variants were validated via Sanger sequencing and correlated…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
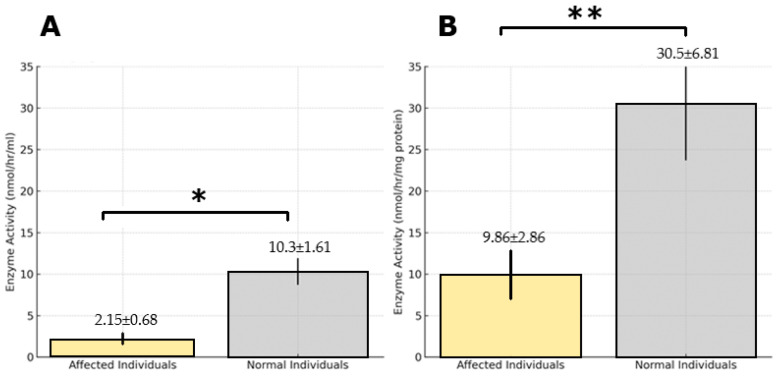 Figure 1
Figure 1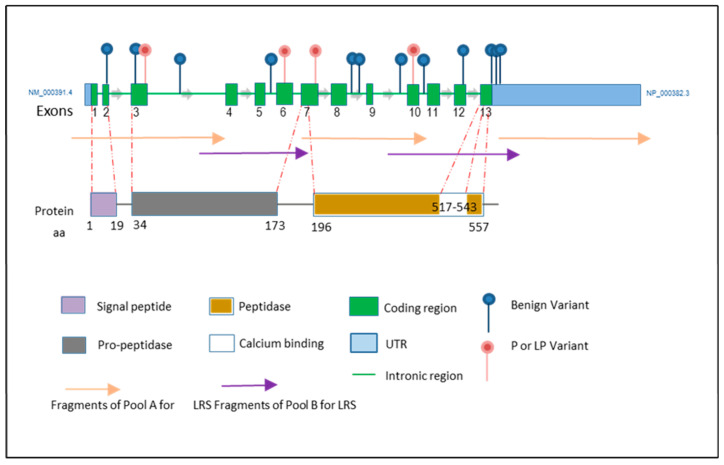 Figure 2
Figure 2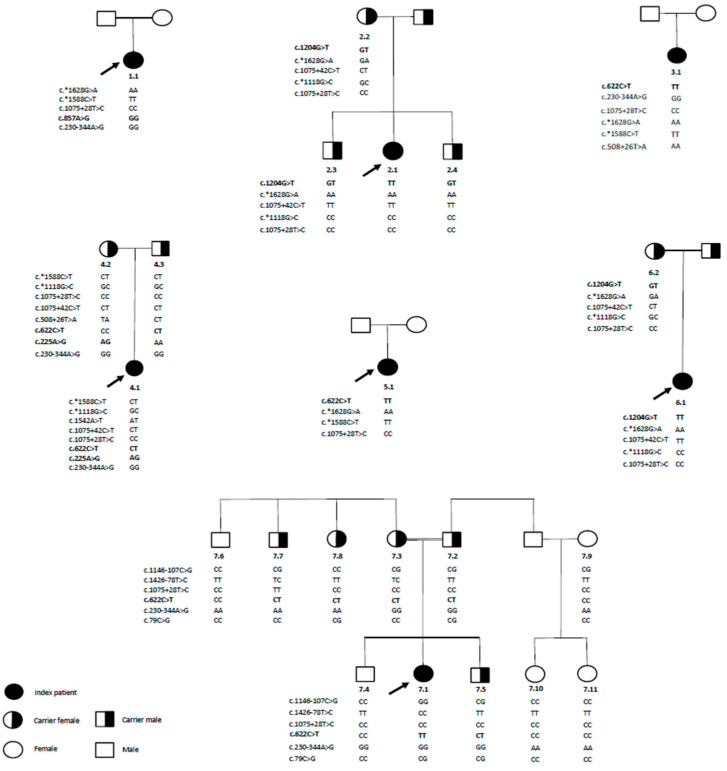 Figure 3
Figure 3Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsLysosomal Storage Disorders Research · Cellular transport and secretion · Signaling Pathways in Disease