nCas9-based method for rolling-circle DNA substrate generation
Nischal Sharma, Kelsey S. Whinn, Harshad Ghodke, Antoine M. van Oijen, Jacob S. Lewis, Lisanne M. Spenkelink

TL;DR
This paper introduces a new method using nCas9 to create customizable rolling-circle DNA substrates for various DNA-related applications.
Contribution
A novel nCas9-based method for generating rolling-circle DNA substrates with precise control over gap size and fork position.
Findings
nCas9 can be programmed to create specific DNA fragments on an 18-kb plasmid.
The method allows for the construction of DNA forks with controlled flap length and gap size.
The generated substrates were successfully used in an in vitro single-molecule DNA replication assay.
Abstract
Rolling-circle DNA replication is a DNA-duplication mechanism whereby circular DNA templates are continuously copied to produce long DNA products. It is widely used in molecular diagnostics, DNA sequencing, nanotechnology, and in vitro DNA replication studies. The efficiency of rolling-circle replication reaction heavily relies on the quality of the rolling-circle DNA template. Existing methods to create rolling-circle DNA substrates often rely on unique restriction sites and have limited control over replication fork topology and position. To address these limitations, we present a straightforward, customizable, and efficient strategy for producing rolling-circle DNA substrates with control over gap size and fork position. Our method relies on the use of nickase Cas9 (nCas9), which can be programmed to target specific DNA sequences using guide RNAs. In a one-pot reaction, we target…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
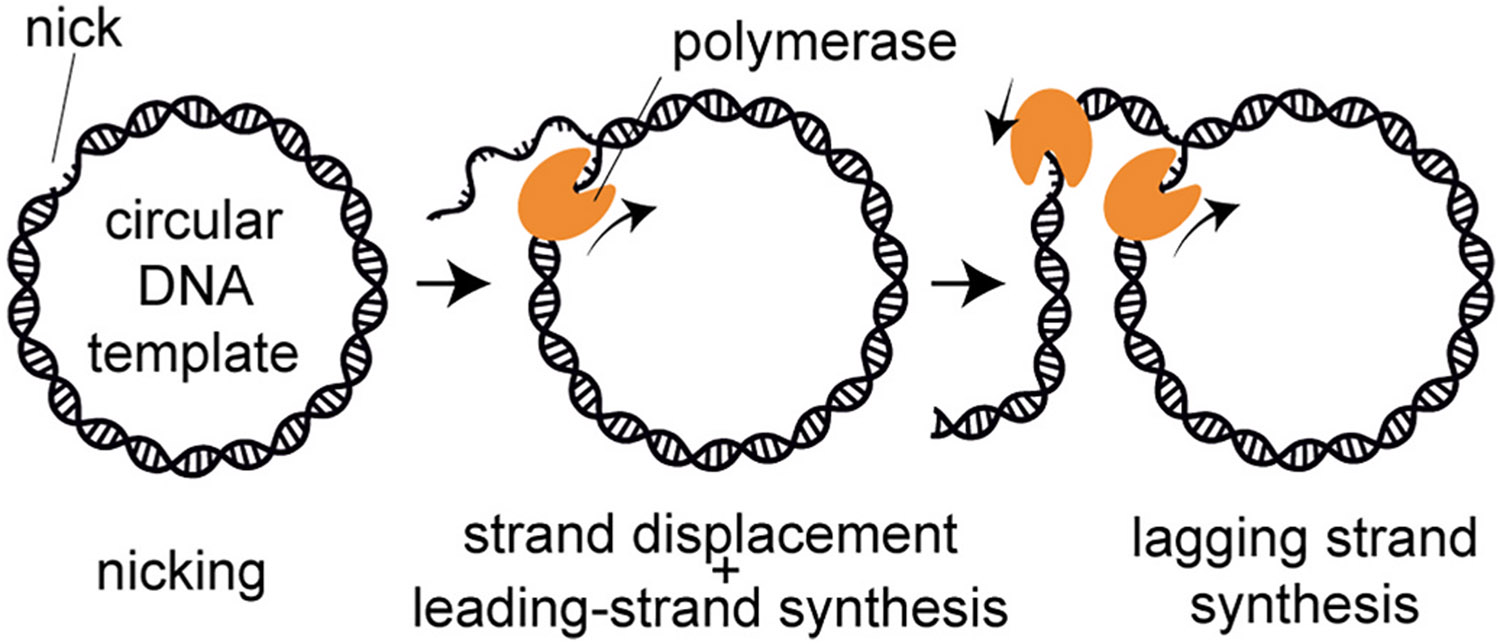 Figure 1
Figure 1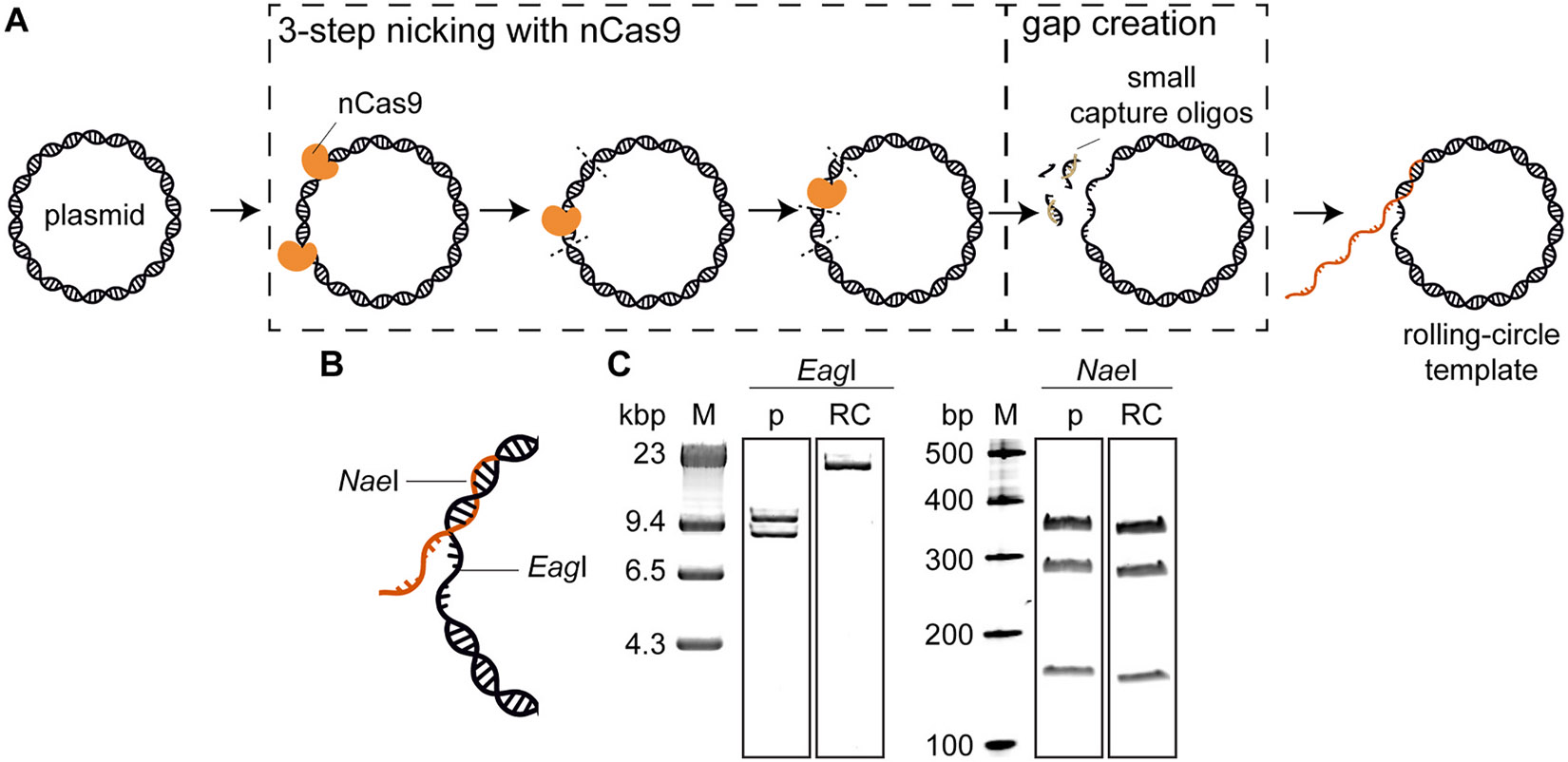 Figure 2
Figure 2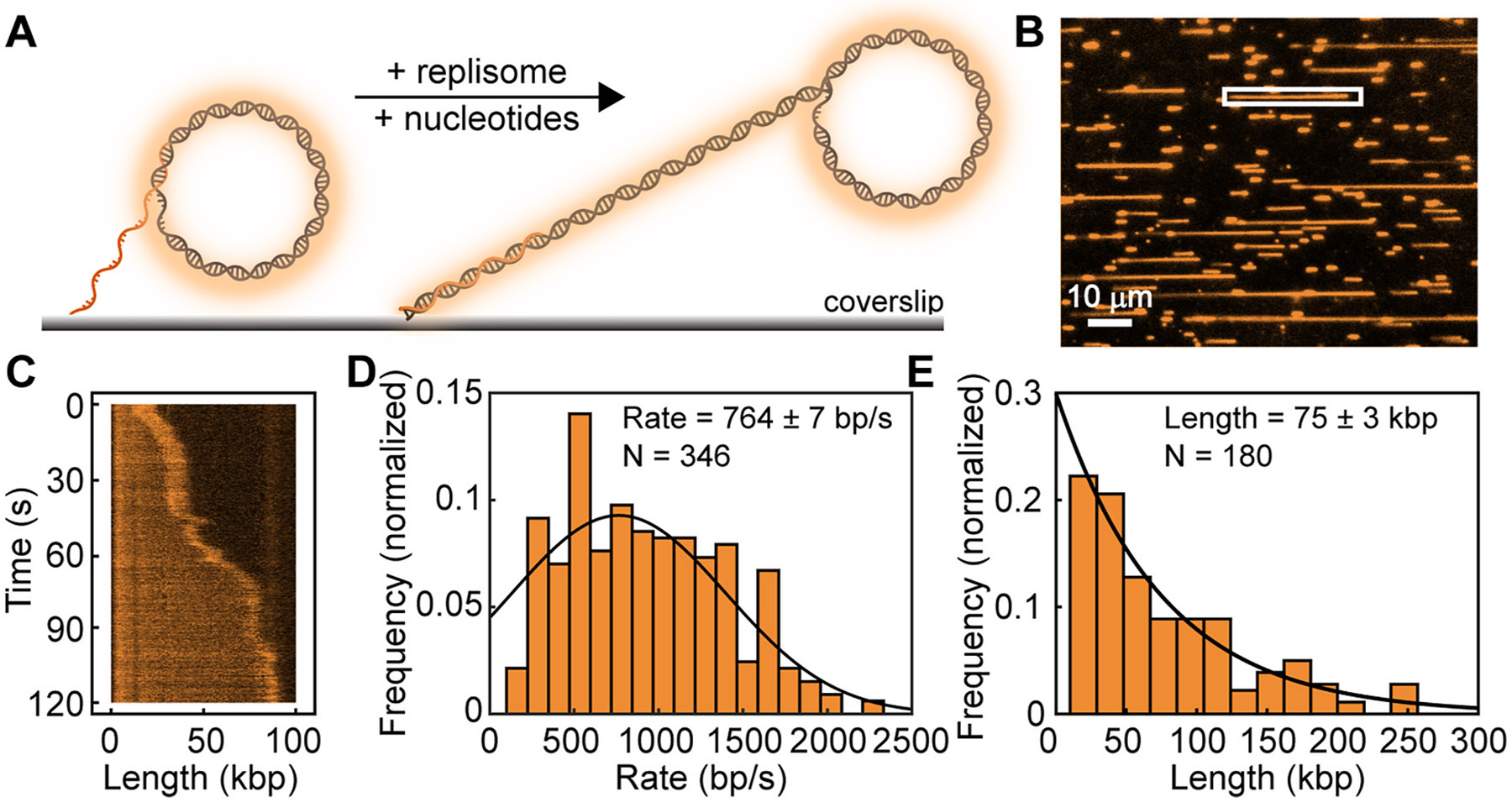 Figure 3
Figure 3Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsAdvanced biosensing and bioanalysis techniques · CRISPR and Genetic Engineering · RNA Interference and Gene Delivery
Introduction
Rolling-circle replication is a mechanism of DNA replication first described in bacteriophage φX174 [1,2]. Through rolling-circle replication, circular DNA templates are continuously copied to produce long DNA products (Fig. 1). In bacteriophages, this process begins with the nicking of one strand of the circular double-strand plasmid [3]. Next, a DNA polymerase binds to the nick and uses the 3′-hydroxyl group as a primer to initiate synthesis. The polymerase uses the intact strand as a template, while displacing the nicked strand as it proceeds around the circle [3,4]. As replication progresses, the continuously synthesized single-stranded DNA strand, the leading strand, is displaced and extruded. This newly-synthesized DNA serves as a template for lagging-strand synthesis in the form of short segments known as Okazaki fragments [4].
Rolling-circle DNA replication is generally processive and isothermal. Therefore, it has been used for molecular diagnostics, DNA sequencing, and nanotechnology [5-7]. Furthermore, rolling-circle replication has played a pivotal role in studying the fundamental mechanisms that underlie DNA replication [3]. The application of rolling-circle replication in single-molecule studies has enabled real-time visualization of individual replicating DNA molecules [8-10] and provided detailed insights on the diverse behaviors of individual replication components from both viruses [11] and prokaryotes [12,13].
The efficiency of rolling-circle replication in these applications requires highly purified undamaged DNA. These templates typically consist of a gapped double-stranded plasmid with a 5′-unpaired single-stranded overhang. Rolling-circle DNA substrates have been created through three main methods. One method starts by annealing a partially-complementary oligonucleotide fork to one end of the single-stranded plasmid [14]. The single-stranded plasmid is then converted to a double-stranded plasmid through primer extension by a DNA polymerase. This method relies on the efficiency of the DNA polymerase and has limited control over the exact gap size. Alternatively, rolling-circle substrates have been created through strand-displacement DNA synthesis at sites of nicks on double-stranded plasmid templates [15]. This method results in substrates lacking a gap at the fork and produce 5′-tails of variable lengths. The final method involves the creation of a single-stranded gap in a double-stranded plasmid, through the use of site-specific nicking enzymes [16]. While this method allows control over the gap size and flap length, it has limited flexibility to modify the fork position. Changing the fork position and gap size requires additional cloning steps to introduce or remove nickase recognition sites.
To address these limitations, we developed a method to create rolling-circle DNA substrates using the nCas9 enzyme, which allows for flexible manipulation of fork position and gap size. nCas9 is a modified version of the Clustered-Regularly-Interspaced-Short-Palindromic-Repeats (CRISPR)-associated protein 9 (Cas9) enzyme that can be programmed to target specific DNA sequences using guide RNAs (gRNAs) [17]. Due to the inactivation of one of its endonuclease domains, nCas9 nicks the DNA, unlike wild-type Cas9, which cuts the DNA. Here, we describe how we produce and validate a rolling-circle DNA template using 18-kb plasmid DNA. We show that this template enables efficient single-molecule rolling-circle replication.
Materials and methods
Reagents
2.1.
Chemicals:
Acetone (Chem-Supply), agarose (Bioline), anti-digoxigenin-AP fab fragments (Life technologies), (3-Aminopropyl) triethoxysilane (ThermoFisher Scientific), ATP (Jena Bioscience), deoxynucleotidetriphosphates (dATP, dCTP, dGTP, dTTP) (Jena Bioscience), biotin-polyethyleneglycol (PEG) and mMPEG (MW 5000) (Laysan Bio), Bovine Serum Albumin (BSA, Sigma-Aldrich), dithiothreitol (DTT) (Astral Scientific), Ethylenediaminetetraacetic acid (EDTA, Sigma-Aldrich), ethanol (Chem-Supply), hydrogen chloride (Sigma-Aldrich), nucleoside triphospates (UTP, CTP, GTP, TTP) (Jena Bioscience), potassium glutamate (Sigma-Aldrich), magnesium chloride (Sigma-Aldrich), magnesium acetate (Sigma-Aldrich), neutravidin (ThermoFisher Scientific), sodium dodecyl sulfate (SDS, Sigma-Aldrich), sodium chloride (Sigma-Aldrich), sodium bicarbonate (Sigma-Aldrich), SYTOX Orange Nucleic Acid Stain (ThermoFisher Scientific), Tris (Astral Scientific), Tween-20 (Sigma-Aldrich).
Enzymes:
Nickase Cas9 (M0650S) (New England Biolabs), T4 DNA ligase (M0202S) (New England Biolabs), NaeI (R0190S) (New England Biolabs), EagI-HF (NEB #R3505) (New England Biolabs).
E. coli DNA replication proteins were purified previously: DnaB6 (DnaC)6 helicase-loader complex [18,19], the Pol III αεθ polymerase core [20], Pol III τ_3_δδ’ψχ clamp loader complex [20], β_2_ sliding clamp [21], single-stranded DNA-binding protein (SSB) [22] and DnaG primase [23].
Buffers:
Blocking buffer (1 × : 50 mM Tris-HCl (pH 7.9), 50 mM KCl, 2 % (v/v) Tween-20), E. coli replication buffer (1 × : 25 mM Tris-HCl (pH 7.9), 10 mM magnesium acetate, 50 mM potassium glutamate, 0.1 mM EDTA, 0.0025 % (v/v) Tween-20, 0.5 mg/mL BSA), Quenching buffer (2 × DNA Gel Loading Dye, 200 mM EDTA, 2 % (w/v) SDS), NEB buffer r3.1 (100 mM NaCl, 50 mM Tris-HCl, 10 mM MgCl2, 100 μg/ml Recombinant Albumin, pH 7.9 at 25 °C), NEB rCutSmart buffer (50 mM Potassium Acetate, 20 mM Tris-acetate, 10 mM Magnesium Acetate, 100 μg/ml Recombinant Albumin, pH 7.9 at 25 °C), TE buffer (10 mM Tris.HCl pH 7.6, 1 mM EDTA, 300 mM NaCl), Tris acetate EDTA buffer (TAE; 40 mM Tris, 20 mM acetic acid, 1 mM EDTA, pH 8.3).
Oligonucleotides
2.2.
All PAGE-purified oligonucleotides were purchased from Integrated DNA Technologies (IDT, USA). The oligonucleotide sequences used for constructing the rolling-circle DNA substrates using one-pot and two-pot reactions are as follows in Tables 1 and 2.
Hybridization of nCas9-gRNAs
2.3.
The hybridization of CRISPR RNA (crRNAs) and Trans-activating CRISPR RNA (tracrRNA) oligos was carried out as per IDT instructions in the following steps.
RNA was resuspended in nuclease-free duplex buffer (IDT) to a final concentration of 100 μM.Equal concentrations of tracrRNA and crRNA were mixed to achieve a final concentration of 15 μM gRNA in nuclease-free duplex buffer. This mixture was heated at 95 °C for 5 min and then allowed to cool to room temperature overnight.Subsequently, equal concentrations of nCas9 and each gRNA were incubated in separate tubes at room temperature for 15 min, forming the different nCas9–gRNA complexes.
While optimisation of crRNA–tracrRNA hybridization conditions is likely possible, the conditions used here gave 100 % nicking efficiency on our template (Fig. 2). Therefore, we did not explore this further.
Construction of the rolling-circle DNA template
2.4.
We targeted nCas9 to create nicks in the plasmid DNA, creating 8–11-nt fragments. These fragments were removed by the addition of a large excess of capture oligos with complimentary sequence, resulting in a gapped plasmid. Finally, the fork oligo is annealed and ligated to create the rolling-circle template. All these steps were carried out in a one-pot reaction, without the need for purification in between steps.
The map of the 18-kbp pUBER plasmid, as previously described by Mueller et al., 2020 (27), and the specific nicking sites for gRNAs to introduce a DNA gap for the attachment of a biotinylated oligo are provided in Supplementary Fig. S1A-B.
Nicking: The nicking reaction was carried out in three sequential steps. First, 50 μg (42 nM) of pUBER plasmid was treated with a 10-fold excess of nCas9–gRNA1 and nCas9–gRNA4 (420 nM each, Table 1) in 1 × NEB r3.1 buffer at 37 °C for 4 h in a thermal cycler, as per IDT instructions. The nicking reaction was then heat-inactivated at 75 °C for 10 min to denature nCas9 enzyme and gRNA molecules (omission of this step results in a reduction of overall efficiency). After allowing the reaction to cool down to room temperature for approximately 15–20 min, the second and third nicking reactions were carried out in the same way using nCas9-gRNA2 and nCas9-gRNA3 (Table 1), respectively. These subsequent reactions were carried out separately to prevent potential interference between nCas9-gRNA complexes.Gapping: A 50-fold molar excess of capture oligo-1 and capture oligo-2 was added to the nicking reaction in step i) and incubated at 80 °C for 20 min to displace the three small DNA fragments located between the two flanking nicked regions of plasmid DNA. Following this, the temperature was slowly decreased at a rate of 1 °C/min until it reached 16 °C to facilitate the annealing of the complementary capture oligos to the displaced oligos.Annealing and ligation of the biotinylated Oligo: A 100-fold excess of partially-complementary biotinylated oligonucleotide (with a 10-nt complementary sequence) was added to the reaction mixture in step ii) and incubated at 70 °C for 15 min. Annealing was achieved by slowly decreasing the temperature to 16 °C at a rate of 1 °C/min. The biotinylated oligonucleotide was ligated by adding 62.5 units/μg T4 DNA ligase, supplemented with 12 mM ATP and 10 mM DTT, once the temperature dropped to 37 °C. The reaction was then incubated at 16 °C for 18 h.Purification: Before purification, the ligase was inactivated by increasing the temperature to 65 °C for 10 min. The final forked DNA substrate was then purified from excess oligos (biotinylated fork oligos and hybridized capture and displaced oligos) using the gel-filtration chromatography as described previously [24]. Briefly, the annealing and ligation reaction from step iv) was loaded onto a Sepharose 4B column (1 × 25 cm; Sigma-Aldrich) equilibrated in gel filtration buffer (10 mM Tris-HCl pH 8.0, 1 mM EDTA, and 300 mM NaCl). The DNA substrate eluted as a single peak in the void volume. The final substrate was stored at −80 °C.
Target binding conditions can likely be optimised, for example by changing the magnesium concentration in the buffer. However, the conditions used here gave us 100 % nicking efficiency, so we did not explore target-binding optimizations. Any off-target nicks that may be introduced, will be re-ligated in subsequent steps and are therefore not of concern.
Verification
2.5.
Correct assembly of the template was verified using the restriction enzymes EagI and NaeI. The restriction digestion reaction was carried out by incubating 30–150 ng of the rolling-circle DNA substrate with 500 units/μg of either EagI or NaeI in 1 × rCutSmart buffer at 37 °C for 2 h. The reactions were quenched by addition of stop solution (40 mM EDTA and 2 % (w/v) SDS) at a volume equal to the reaction volume.
DNA products from the EagI digestion reactions, which generate larger DNA fragments, were separated using a 0.6 % (w/v) agarose gel in 1 × TBE buffer at 15 V for 800 min in a Wide Mini-Sub Cell GT System (Bio-Rad). The smaller DNA fragments generated by NaeI digestion were resolved on a 4–20 % Mini-PROTEAN TGX precast protein gel (Bio-Rad) in 1 × TBE buffer at 150 V for 75 min, using a Mini-PROTEAN Tetra vertical electrophoresis cell (Bio-Rad) (Refer to Supplementary Fig. S1A for the number of DNA fragments generated after EagI and NaeI digestion reactions).The SYBR-Gold-stained gels were imaged on an Amersham Typhoon biomolecular imager (Cytiva).
If these verification steps indicate that the substrate was not formed correctly, optimisation of the gRNAs, the gapping, or the ligation might be required (see Supplementary Table 1 for more detail).
Single-molecule rolling-circle replication assay
2.6.
To show that our DNA substrate can support DNA replication, we carried out in vitro single-molecule rolling-circle replication assays. Replication reactions were carried in microfluidic flow cells assembled using a Polydimethylsiloxane (PDMS) flow chamber placed on top of a glass coverslip functionalized with streptavidin-PEG-biotin, as previously described [12,14,18,24-27]. After assembly, we introduced with 300 μL of blocking buffer and incubated for at least 10 min to prevent nonspecific interactions of proteins and DNA with the flow-cell surfaces. The flow cell was then washed with 300 μL of 1 × replication buffer to remove the blocking buffer.
Single-molecule rolling-circle replication assays were carried out as describes previously [12], Briefly, 30 pM of rolling-circle DNA substrates was incubated with 15 nM DnaB_6_ (DnaC)6 in degassed 1X replication buffer in the presence of 1 mM ATP and 10 mM DTT at 37 °C for 2 min. Then, the DNA intercalating dye, SYTOX Orange (150 nM), was added and the reaction mixture was loaded into the flow cell at a constant rate of 10 μL/min to tether the DNA to the glass microscope coverslip via biotin–streptavidin–biotin linkage. Once an appropriate surface density of DNA was achieved, replication was initiated by loading of 30 nM αϵθ, 10 nM τ3δδ′ψχ, 46 nM β2, 75 nM DnaG, and 20 nM SSB, in 1X replication buffer, with 1 mM ATP, 10 mM DTT, 250 μM of each NTP, 50 μM of each dNTP, and 150 nM SYTOX Orange, at a constant flow of 20 μl/min.
Single-molecule imaging conditions
2.7.
Single-molecule experiments were carried out using an Eclipse Ti-E inverted fluorescence microscope (Nikon, Japan) equipped with a CFI Apo TIRF 100× oil-immersion objective (NA 1.49, Nikon, Japan), as described previously [12,28,29]. The temperature of the flow cells was maintained at 31.2 °C using an electrically heated chamber (Okolab, USA). Images were captured using a 512 × 512 pixel^2^ EM-CCD camera (Andor iXon Life, EMCCD). NIS-Elements software (Nikon, Japan) was used to control the microscope, and the focus was maintained using the Perfect Focus System (Nikon, Japan). The DNA molecules were stained with 150 nM SYTOX Orange and imaged using a 532-nm laser (Coherent, Sapphire 532–200 CW) at 0.1 mW/cm^2^. The typical acquisition parameters were set at 1 frame/s for 4 min with an exposure time of 200 ms.
Analysis of single-molecule replication kinetics
2.8.
Image analysis was carried out in ImageJ, v1.51w. First, raw acquisitions in nd2 format were corrected for beam profile and x,y drift. Next, replicating molecules of interest were selected and a kymograph, a graphical representation that shows the progression of replication forks along the DNA template over time, was generated for each molecule.
Rate analysis:
Here, replication rate refers to the speed at which the DNA molecule grows (e.g. the speed at which replication progresses). Positions of the tip of the replicating molecule were determined as previously described [25]. These coordinates were used to detect individual segments of constant rate. Finally, the data were fit to a Gaussian distribution function using MATLAB 2016b.
DNA length analysis:
Here, the DNA length refers to the total length of the replicated DNA product, after 2 min of imaging. The length of the DNA product was determined by measuring the final length of the 1D line (in pixels). Finally, the data were fit to a single exponential function using MATLAB 2016b.
Results and discussion
Template construction
3.1.
We developed a method to generate rolling-circle templates using nCas9. First, the plasmid DNA is nicked in three steps to introduce four nicks (Fig. 2A). We add nCas9–gRNA complexes to create nicks at four locations (Fig. 2A, Supplementary Fig. S1B). The nicks at these four locations create three small DNA fragments. These small fragments are then displaced from the plasmid by increasing the temperature, before they are captured by adding complementary capture oligos (Fig. 2A). This process creates a 35-nt-long DNA gap on the plasmid, where the partially-complementary biotinylated fork oligonucleotide can be attached (Fig. 2A, Supplementary Fig. S1B). We achieve an overall efficiency of 47 ± 4 % (N = 3).
Validation
3.2.
To verify correct assembly of the rolling-circle templates, we used two restriction enzymes to cut the template. EagI is used to verify the presence of the gap (Fig. 2B). EagI has two recognition sites within pUBER plasmid (Supplementary Fig. S1A). Therefore, restriction of pUBER with EagI will result in two linear fragments (~8.5 kb and ~9 kb, respectively). For successfully-formed rolling-circle substrates, however, one recognition site falls within the gap and cannot be cleaved. Therefore, restriction of the correctly-formed rolling-circle substrate with EagI is expected to produce an 18-kb linear fragment (Fig. 2B. Supplementary Fig. S1B). Fig. 2C (left) shows that treatment with EagI produces one linear fragment, indicating all the templates contain a gap. NaeI was used to confirm successful ligation of the fork oligonucleotide to the DNA substrate (Fig. 2B). NaeI cleaves the plasmid DNA and the rolling-circle template at five sites (Supplementary Fig. S1A), resulting in five fragments (~12 kb, 6 kb, 354 bp, 283 bp 160 bp). If, however, the fork is not successfully annealed and ligated, one of these sites is located in the gap and will not be cut (Supplementary Fig. S1B). As a result, digestion of unsuccessfully-created substrates with NaeI would not show the 160-bp and 354-bp bands. Fig. 2C (right) shows the three lowest bands of the NaeI-restricted plasmid and rolling-circle template on a precast gel. All three bands are present, indicating successful annealing and ligation of the fork oligo.
Single-molecule visualization
3.3.
To demonstrate the use of this template, we chose a single-molecule rolling-circle assay using the E. coli DNA replication system. Comprehensive analysis of this reaction has been done previously [8,25,26,30-36]. We simply use this system to demonstrate that our substrate supports replication with similar efficiencies to other rolling-circle templates. We loaded our template onto the surface of a microscope coverslip in a microfluidic flow cell in the presence of SYTOX Orange. We then added all the purified E. coli replisome components to initiate replication. The laminar flow of buffer will stretch the newly-synthesized DNA in the direction of flow (Fig. 3A) allowing visualization though SYTOX Orange. Fig. 3B shows a typical field of view, where each line represents a replication product. Our rolling-circle template has a replication efficiency (percent of templates that are replicated) of 44 ± 4 %, similar to efficiencies reported for other substrates [19]. We can monitor replication of individual templates over time (Fig. 3C), allowing us to measure the instantaneous rate of replication [25] and the final product length after 2 min for all molecules. The rate (764 ± 7 bp/s) and product length (75 ± 3 kbp) are the same as previously reported rates for rolling-circle replication by the E. coli replisome [34].
Conclusion
In summary, we present a straightforward, customizable, and efficient strategy to generate rolling-circle DNA substrates with direct control over the gap size and fork position (Table 2). Our method has comparable cost and time requirements to existing approaches (Table 2). We show that these template support robust DNA replication, with similar efficiencies and rates to other previously reported rolling-circle templates.
Gap size is known to affect the efficiency of replisome assembly [45]. Our method can be used to produce optimal gap sizes for different replisomes. Control over the fork position can be used to provide spatial separation between the fork and a genetic element of interest, such as transcription promotors or site-specific DNA damage [12]. These templates, therefore, allow studies on the behavior of the replisome upon encountering obstacles like secondary DNA structures, DNA-bound proteins, and lesions. Furthermore, control over the fork position enables the deliberate avoidance or targeting of regions with high secondary structure. While it is known that secondary structures can affect nCas9 nicking efficiency, methods have been developed to overcome these issues [46]. Thus, we anticipate that our method will allow production of rolling-circle templates for the study of replication protein dynamics on increasingly complex and crowded DNA substrates [47].
Supplementary Material
Supplementary Information
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Schröder CH, Erben E, Kaerner HC, A rolling circle model of the in vivo replication of bacteriophage phi X 174 replicative form DNA: different fate of two types of progeny replicative form, J. Mol. Biol 79 (4) (1973) 599–613.4585979 10.1016/0022-2836(73)90066-1 · doi ↗ · pubmed ↗
- 2Gilbert W, Dressler D, DNA replication: the rolling circle model, Cold Spring Harbor Symp. Quant. Biol 33 (1968) 473–484.4891987 10.1101/sqb.1968.033.01.055 · doi ↗ · pubmed ↗
- 3Ruiz-Maso JA, , Plasmid rolling-circle replication, Microbiol. Spectr 3 (1) (2015). Plas-0035-2014.10.1128/microbiolspec.PLAS-0035-201426104557 · doi ↗ · pubmed ↗
- 4Lee J, , Coordinated leading and lagging strand DNA synthesis on a minicircular template, Mol Cell 1 (7) (1998) 1001–1010.9651583 10.1016/s 1097-2765(00)80100-8 · doi ↗ · pubmed ↗
- 5Wu HC, , DNA sequencing using rolling circle amplification and precision glass syringes in a high-throughput liquid handling system, Biotechniques 34 (1) (2003) 204–207.12545561 10.2144/03341 pf 01 · doi ↗ · pubmed ↗
- 6Liu J, , Recent applications of rolling circle amplification in biosensors and DNA nanotechnology, Tr AC, Trends Anal. Chem 160 (2023) 116953.
- 7Zhao W, , Rolling circle amplification: applications in nanotechnology and biodetection with functional nucleic acids, Angew Chem. Int. Ed. Engl 47 (34) (2008) 6330–6337.18680110 10.1002/anie.200705982 · doi ↗ · pubmed ↗
- 8Tanner NA, , Real-time single-molecule observation of rolling-circle DNA replication, Nucleic Acids Res. 37 (4) (2009) e 27.19155275 10.1093/nar/gkp 006PMC 2651787 · doi ↗ · pubmed ↗