Hereditary pulmonary alveolar proteinosis in a 5-year-old child: Diagnostic insights and therapeutic approach
Dhiran Sivasubramanian, Karthick Balasubramanian, Sathwik Sanil, Smrti Aravind, Virushnee Senthilkumar

TL;DR
A 5-year-old child was diagnosed with a rare lung disease using imaging, lab tests, and genetic analysis, and improved after a lung-washing treatment.
Contribution
This case highlights the diagnostic process and treatment of hereditary pulmonary alveolar proteinosis in a pediatric patient.
Findings
The patient showed hypoxemia and a 'crazy-paving' HRCT pattern consistent with PAP.
Genetic testing confirmed a homozygous mutation in the CSF2RA gene, confirming hPAP.
Therapeutic whole lung lavage led to significant clinical improvement.
Abstract
Hereditary pulmonary alveolar proteinosis (hPAP) is a rare disorder caused by mutations in the CSF2RA or CSF2RB genes, leading to impaired surfactant clearance by alveolar macrophages and subsequent respiratory dysfunction. A 5-year-old female with a 2-year history of poor weight gain, fatigue, and intermittent fever was evaluated. Clinical evaluation revealed hypoxemia, while high-resolution computed tomography (HRCT) of the chest showed the characteristic “crazy-paving” pattern suggestive of PAP. Bronchoalveolar lavage (BAL) yielded milky fluid with periodic acid-Schiff (PAS)-positive material, and genetic testing confirmed a homozygous mutation in the CSF2RA gene, consistent with hPAP. The patient underwent therapeutic whole lung lavage (WLL), resulting in significant clinical improvement. This case underscores the challenges of diagnosing pediatric hPAP and the value of integrating…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
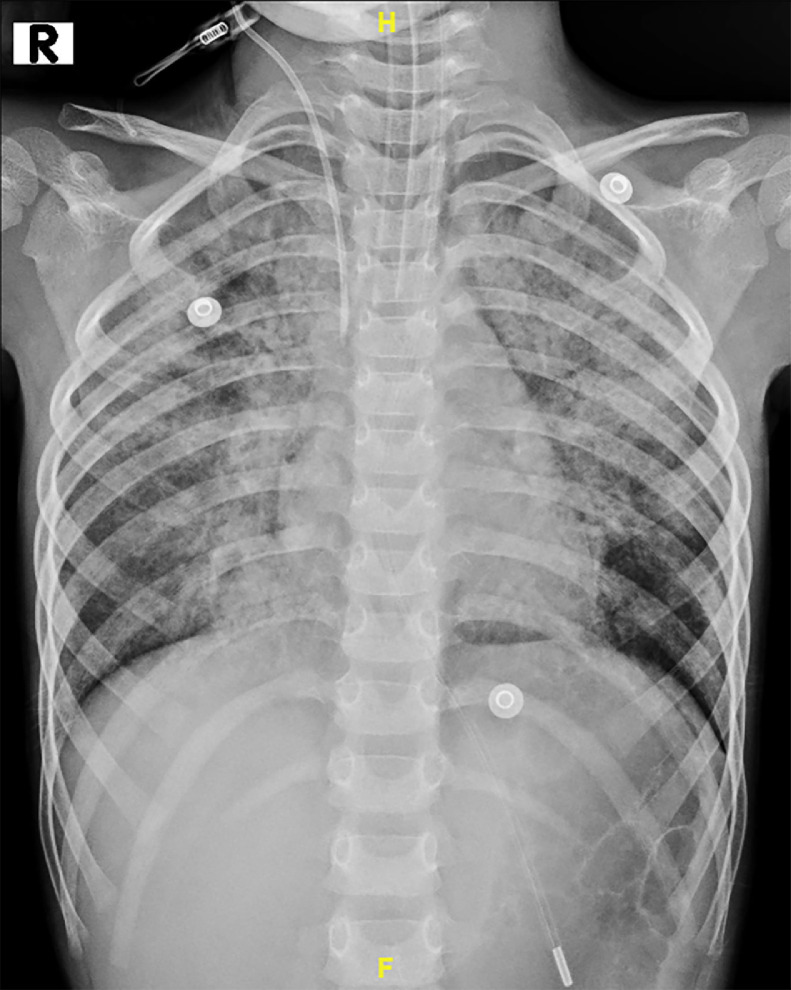 Figure 1
Figure 1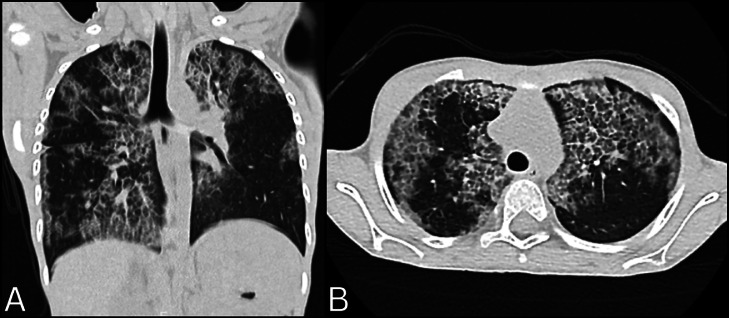 Figure 2
Figure 2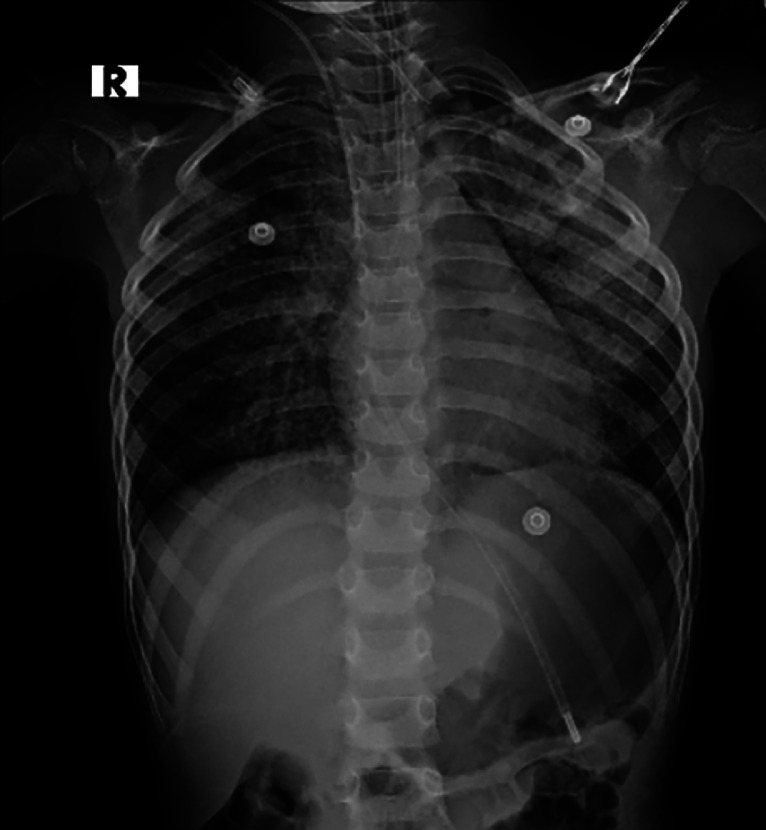 Figure 3
Figure 3Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsNeonatal Respiratory Health Research · Congenital Diaphragmatic Hernia Studies · Medical Imaging and Pathology Studies
Introduction
Pulmonary alveolar proteinosis (PAP) is a rare and potentially fatal form of interstitial lung disease, which is characterized by the accumulation of lipoproteinaceous material, mainly composed of surfactant phospholipid and apoproteins, in the alveoli. This leads to impaired gas exchange and progressive respiratory insufficiency. It results from a disruption in the clearance of surfactant by alveolar macrophages, which normally regulates surfactant homeostasis in the lungs [[1], [2], [3]]. Based on the etiopathogenesis, PAP is classified into three major types: 1) Autoimmune PAP (primary)—the most common form accounting for about 90% of cases, 2) Secondary PAP—occurs as a consequence of underlying conditions such as malignancies, inhalational exposure or immunodeficiencies, which impair macrophage function, 3) Hereditary PAP (hPAP)—a rare genetic form caused by mutations in the CSF2RA or CSF2RB genes, leading to impaired surfactant clearance by alveolar macrophages [1].
Clinically, hPAP often presents in infancy or early childhood with nonspecific respiratory symptoms, including progressive dyspnea, cough, and hypoxemia. Radiologically, high-resolution computed tomography (HRCT) often suggests the diagnosis [1]. But diagnosis requires confirmation through genetic testing, a bronchoalveolar lavage (BAL), or a lung biopsy in select cases [4].
The management of hPAP remains challenging, and it is primarily supportive. Whole lung lavage (WLL) is the mainstay therapy [1,4,5].
We present a case of hereditary pulmonary alveolar proteinosis in a child, aiming to highlight the diagnostic challenges and management considerations associated with hPAP.
Case report
A 5-year-old female was brought to the outpatient clinic with complaints of poor weight gain and increased fatigue while playing with her peers, symptoms she has been experiencing for the past two years. Her parents also reported intermittent episodes of high-grade fever, occurring every three months and lasting about 10 days, during this period. Additionally, they observed that she was shorter than her peers. She had multiple hospitalizations since the age of 3 for recurrent fevers and pneumonia, which were treated with intravenous antibiotics. She was born to parents who were married consanguineously in the second degree, and her antenatal history was unremarkable. All developmental milestones were achieved on time, and her vaccinations were up to date. Her diet was normal, with adequate protein and calorie intake. There was no family history of any similar illnesses.
Upon presentation, her vital signs were as follows: blood pressure of 90/60 mmHg, pulse rate of 100 beats per minute, respiratory rate of 25 breaths per minute, and an oxygen saturation of 95%. The physical examination was normal, with no abnormalities noted in the cardiovascular, respiratory, or central nervous systems. Routine blood tests were within reference range (Table 1). Arterial blood gas analysis showed hypoxemia with a PaO_2_ of 34 mmHg, a PaCO_2_ of 41 mmHg, and a pH of 7.36. A Chest radiograph (X-ray) demonstrated bilateral, diffuse, non-homogeneous opacities (Fig. 1). Followed by a high-resolution computed tomography (HRCT) of the chest that showed large, patchy areas of ground-glass opacities with interlobular septal thickening across all lobes of both lungs, resulting in a classic “crazy-paving” pattern (Fig. 2). Additionally, there was mild involvement of the right upper paratracheal and subcarinal lymph nodes, measuring up to 3 mm. The complete antinuclear antibody (ANA) panel was negative.Table 1. Routine blood investigations.Table 1. InvestigationPatient valueReference valueHemoglobin (Hb)14 g/dL12-15 g/dLMean corpuscular volume (MCV)82.1 fL80-100 fLPlatelet count327,000 cells/µL150,000-450,000 cells/µLTotal white blood cell count (WBC)5,800 cells/μL4,500-11,000 cells/μLTotal bilirubin0.37 mg/dL0.3-1.2 mg/dLDirect bilirubin0.10 mg/dL0-0.3 mg/dLSerum albumin5.0 g/dL3.5-5 g/dLAlanine transaminase (ALT)44 IU/L5-44 IU/LAspartate aminotransferase (AST)18 IU/L5-35 IU/LAlkaline phosphatase (ALP)315 U/L273.47-871.44 U/LUrea39 mg/dL7-40 mg/dLCreatinine0.34 mg/dL0.5-1.2 mg/dLBicarbonate21 mmol/L22-29 mmol/LSodium138 mmol/L136-145 mmol/LPotassium4.3 mmol/L3.5-5.1 mmol/LFig. 1Chest radiograph (X-ray) showing bilateral diffuse nonhomogeneous opacities.Fig 1. Fig. 2High-resolution computed tomography (HRCT) (A) coronal and (B) axial view of the chest showing large patchy areas of ground glass opacities with interlobular septal thickening involving all lobes of bilateral lung parenchyma, resulting in a crazy-paving pattern.Fig 2
Given the radiological suspicion of PAP, the patient underwent bronchoscopy with bronchoalveolar lavage (BAL) of the right lower lobe. The BAL fluid appeared milky and turbid, becoming increasingly dense with each lavage. Periodic acid-Schiff (PAS) staining of the fluid was positive, with the granular material consistent with alveolar proteinosis. The total white blood cell count in the BAL fluid was 360/mm³, with 28% lymphocytes and 65% macrophages. Granulocyte-macrophage colony-stimulating factor (GM-CSF) autoantibodies were not detected in either the serum or the BAL fluid. Genetic testing revealed a homozygous mutation in the CSF2RA gene, confirming the diagnosis.
The patient underwent therapeutic whole-lung lavage (WLL) under general anesthesia, with successful fluid clearance achieved after 15 cycles. A chest radiograph taken a day after the procedure showed a significant reduction in the opacities over the right lung (Fig. 3) in comparison to (Fig. 1). The procedure was repeated a few more times over the month. At a follow-up visit the next month, she had a significant improvement in her symptoms.Fig. 3. Chest radiograph (X-ray) post right whole lung lavage (WLL) showing reduction of the opacities in the right lung in comparison to the first X-ray in Fig. 1.Fig 3
Discussion
Pulmonary alveolar proteinosis is rare in children, with an estimated incidence of two cases per million under the age of 18 [6]. While the majority of them are autoimmune PAP (primary), hereditary PAP is extremely rare, accounting for less than 6% of these cases [3]. The CSF2RA or CSF2RB genes encode the alpha and beta subunits of the granulocyte-macrophage colony-stimulating factor (GM-CSF) receptor, respectively [1]. These mutations disrupt surfactant homeostasis by impairing the alveolar macrophages' ability to clear surfactant, leading to accumulation in the alveoli and resulting in respiratory dysfunction [2,3].
The patient presented with nonspecific symptoms that had persisted over two years. These symptoms, along with a history of recurrent pneumonia and consanguineous parentage, suggested a possible underlying genetic disorder. Routine blood investigations were largely unremarkable, except for hypoxemia. However, the imaging findings, particularly the HRCT scan, were crucial in establishing the diagnosis. The initial chest radiograph showed diffuse, non-homogeneous opacities—a non-specific finding often seen in various pulmonary pathologies [3]. Although a chest radiograph can serve as an initial screening tool [3], it was the HRCT that revealed the hallmark “crazy-paving” pattern of PAP [1,7]. This pattern is named for the resemblance of ground-glass opacities superimposed with interlobular septal thickening to a pathway paved with irregularly broken pieces of stone [7]. Typically, the opacities are bilateral and diffuse, often involving the perihilar and basal lung regions, as seen in this child. Lymphadenopathy, although less common, can be observed in PAP and is more frequently seen in secondary forms. It may reflect a reactive process or an association with infections due to impaired pulmonary defense mechanisms [8].
The “crazy-paving” pattern can also be seen in other conditions, such as cardiogenic pulmonary edema, acute respiratory distress syndrome (ARDS), pneumocystis pneumonia (PCP), and various neoplastic processes [9,10]. Therefore, the diagnosis must be confirmed with additional tests, including BAL (the gold standard) and genetic studies in hereditary cases. In the appropriate clinical context, particularly in pediatric cases with unresolved respiratory symptoms and growth concerns, this pattern should raise strong suspicion for PAP.
The milky appearance of the BAL fluid, along with positive PAS staining, confirmed our diagnosis of PAP. The absence of GM-CSF autoantibodies excluded the autoimmune form of PAP, while genetic testing identified a homozygous mutation in the CSF2RA gene, confirming the hereditary form [9]. This distinction is crucial for diagnosis and treatment, as recombinant GM-CSF therapy is effective in autoimmune PAP but unlikely to work in hPAP due to the underlying receptor dysfunction [9,11].
The management of hPAP is primarily supportive, with WLL being the cornerstone of therapy. WLL involves the physical removal of accumulated surfactant material from the alveoli, improving gas exchange and respiratory function [12]. However, WLL is not a cure, and patients often require repeated procedures [9,12]. The long-term prognosis of hPAP varies, and some patients may develop progressive respiratory failure despite treatment. In such cases, lung transplantation may be considered [12].
The genetic basis of hPAP raises important considerations for family counseling and screening. In this case, the consanguineous parents increased the likelihood of an autosomal recessive disorder such as hPAP. Genetic counseling should be offered to the family, and siblings should be screened for the CSF2RA mutation, even if they are asymptomatic [13]. Early diagnosis and intervention in affected siblings can improve outcomes and prevent complications.
Conclusion
This case highlights the diagnostic challenges associated with hPAP in children and emphasizes the importance of a systematic approach to evaluation, including clinical, radiological, histopathological, and genetic assessments. HRCT remains essential in the radiological evaluation of PAP. Recognizing the “crazy-paving” pattern in children with chronic respiratory symptoms should prompt consideration of hereditary PAP, necessitating a comprehensive diagnostic approach involving bronchoscopy and genetic testing. While WLL remains the primary treatment, further research is needed to develop targeted therapies for the underlying genetic defects in hPAP.
Patient consent
Written informed consent for publication of this case was obtained from the patient.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Salvaterra E.Campo I.Pulmonary alveolar proteinosis: from classification to therapy Breathe (Sheffield, England)162202020001810.1183/20734735.0018-2020 PMC 734161632684997 · doi ↗ · pubmed ↗
- 2Rosen S.H.Castleman B.Liebow A.A.Pulmonary alveolar proteinosis New Eng J Med 2582319581123114210.1056/NEJM 19580605258230113552931 · doi ↗ · pubmed ↗
- 3Suzuki T.Sakagami T.Young L.R.Carey B.C.Wood R.E.Luisetti M.Hereditary pulmonary alveolar proteinosis: pathogenesis, presentation, diagnosis, and therapy Am J Respirat Critical Care Med 1821020101292130410.1164/rccm.201002-0271 OC 20622029 PMC 3001266 · doi ↗ · pubmed ↗
- 4Suzuki T.Trapnell B.C.Pulmonary alveolar proteinosis syndrome Clinic Chest Med 373201643144010.1016/j.ccm.2016.04.006PMC 590218727514590 · doi ↗ · pubmed ↗
- 5Huaringa A.J.Francis W.H.Pulmonary alveolar proteinosis: a case report and world literature review Respirol Case Rep 462016 e 0020110.1002/rcr 2.201PMC 516728628031836 · doi ↗ · pubmed ↗
- 6Mc Carthy C.Avetisyan R.Carey B.C.Chalk C.Trapnell B.C.Prevalence and healthcare burden of pulmonary alveolar proteinosis Orphanet J Rare Dis 131201812910.1186/s 13023-018-0846-y 30064481 PMC 6069872 · doi ↗ · pubmed ↗
- 7Lee C.H.The crazy-paving sign Radiology 2433200790590610.1148/radiol.243304183517517945 · doi ↗ · pubmed ↗
- 8Ishii H.Tazawa R.Kaneko C.Saraya T.Inoue Y.Hamano E.Clinical features of secondary pulmonary alveolar proteinosis: pre-mortem cases in Japan Eur Respirat J 372201146546810.1183/09031936.0009291021282812 · doi ↗ · pubmed ↗