A Comprehensive Approach to Differentiating Systemic Lupus Erythematosus From Other Autoimmune Diseases Presenting With Optic Neuritis
Jason Peng, Nada Alrifai, Salvatore D DeSimone, Pietro M Gentile, Pamela Traisak, Marissa Karpoff, David Feinstein, Hala Eid, Joseph D DeSimone

TL;DR
This paper reviews how optic neuritis can help distinguish systemic lupus erythematosus from other autoimmune diseases like multiple sclerosis and neuromyelitis optica.
Contribution
The study provides a systematic review of diagnostic approaches to differentiate SLE from other autoimmune diseases using optic neuritis as a key indicator.
Findings
SLE-associated optic neuritis is rare but distinct, often bilateral with severe visual impairment and pain.
Recovery of visual acuity is less common in SLE-associated ON compared to multiple sclerosis and idiopathic ON.
Diagnostic differentiation relies on antibody profiles, MRI findings, and blood biomarkers like semaphorins and complement levels.
Abstract
Systemic lupus erythematosus (SLE) is a complex autoimmune disease with significant morbidity and mortality. Early diagnosis is crucial for effective management. Ocular manifestations, particularly optic neuritis (ON), have emerged as potential early signs of SLE. This systematic literature review analyzes 23 studies, including eight retrospective studies, five cross-sectional studies, two cohort studies, four case reports, one narrative review, and three systematic literature reviews, to investigate the use of ON in diagnosing SLE and differentiating it from other autoimmune diseases, most notably multiple sclerosis (MS) and neuromyelitis optica spectrum disorder (NMOSD). A comprehensive literature search was conducted by combining various keywords and MeSH terms, focusing on autoimmune diseases, ON, MS, NMOSD, and SLE. The final selection of 23 studies reflected a methodologically…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
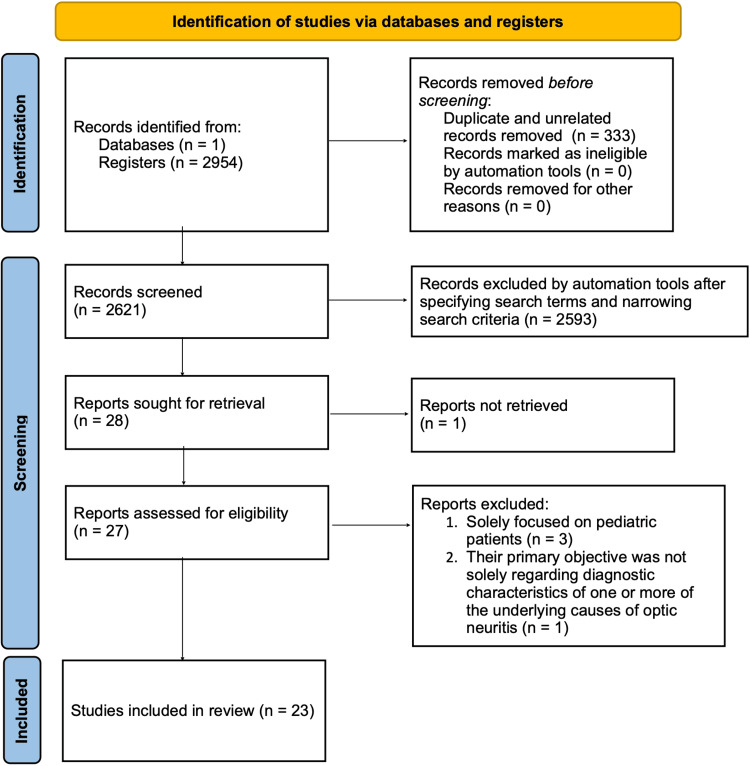 Figure 1
Figure 1| Author | Year | Country | Study type | Population |
|
Zhang et al. [ | 2020 | China | Retrospective study | 225 |
|
Magro Checa et al. [ | 2013 | The Netherlands | Review | N/A |
|
Zhao et al. [ | 2016 | China | Retrospective study | 22 |
|
Nardone et al. [ | 2015 | Austria | Case report | 1 |
|
Kampylafka et al. [ | 2013 | Greece | Cohort study | 370 |
|
Fang et al. [ | 2020 | Taiwan | Retrospective study | 5,488 |
|
Siegel et al. [ | 2022 | USA | Retrospective study | 139 |
|
Martín-Nares et al. [ | 2019 | Mexico | Retrospective study | 12 |
|
Takahashi et al. [ | 2012 | Japan | Cross-sectional study | 47 |
|
Asgari et al. [ | 2013 | Denmark | Review | 1,493 |
|
Bibic et al. [ | 2019 | Canada | Case report | 2 |
|
Caroline Breis et al. [ | 2020 | Brazil | Systematic review | N/A |
|
Jain et al. [ | 2016 | India | Retrospective study | 64 |
|
Thabah et al. [ | 2019 | India | Case report | 8 |
|
Park et al. [ | 2014 | Korea | Retrospective study | 106 |
|
Xiao et al. [ | 2016 | China | Retrospective study | 64 |
|
Bruschi et al. [ | 2020 | Italy | Narrative review | N/A |
|
Sinnecker et al. [ | 2012 | Germany | Cross-sectional study | 28 |
|
Martin et al. [ | 2015 | USA | Case report | 434 |
|
Liang et al. [ | 2019 | China | Cross-sectional study | 1,715 |
|
Ostendorf et al. [ | 2019 | Germany | Cross-sectional study | 75 |
|
Moraitis et al. [ | 2019 | London | Cross-sectional study | 90 |
|
de Souza et al. [ | 2012 | Brazil | Cohort study | 36 |
| Feature | SLE-associated ON | NMOSD-associated ON | MS-associated ON |
| Laterality |
Severe visual impairment, typically bilateral; some unilateral cases as thrombotic ON with antiphospholipid antibodies [ |
Bilateral simultaneous or sequential ON occurring rapidly one after another [ |
Typically unilateral [ |
| Visual recovery |
~50% recover to better than 20/25 visual acuity [ | Not specified |
~87% recover to better than 20/25 visual acuity [ |
| Pain |
Intense; exacerbated by eye movement [ | Not mentioned |
Mild pain upon eye movement [ |
| Associated features |
May involve the optic chiasm; thrombotic ON associated with antiphospholipid antibodies [ |
Coexistence with SLE reported; diagnosis confirmed with MRI, ON, TM, and elevated AQP4 antibodies [ |
Mild disc swelling, diminished color vision, and afferent pupillary defect [ |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsSystemic Lupus Erythematosus Research · Multiple Sclerosis Research Studies · Peripheral Neuropathies and Disorders
Introduction and background
Systemic lupus erythematosus (SLE) is a multisystem autoimmune disease with unknown etiology that causes widespread morbidity and mortality [1]. Because of its complex disease pathogenesis and heterogeneity of symptoms, it is difficult to diagnose and is not a reportable disease, making it expensive to accurately document for epidemiologic studies [1,2]. The global incidence of SLE is 5.15 (1.4-15.13) per 100,000 person-years. Mortality is caused by either organ failure, infection, or cardiovascular disease [2,3]. It takes an individual an average of two years to be diagnosed with SLE after the initial onset of symptoms [4,5]. The timeliness of an SLE diagnosis significantly impacts prognosis, as early detection can avert severe tissue damage [4]. This is especially true considering SLE progression often occurs before the onset of clinical symptoms [5]. This diagnostic delay is clinically significant, as early detection is critical to preventing irreversible organ damage and improving long-term outcomes [4,5]. This is especially true considering SLE progression often begins before overt clinical symptoms appear [5]. Previous studies have shown that ocular manifestations can emerge as a first sign of disease in up to one-third of SLE patients [6]. Among these manifestations, optic neuritis (ON), keratoconjunctivitis sicca, and episcleritis are commonly presenting symptoms of SLE progression [7-9]. ON is relatively rare in the context of SLE, occurring in just 1% of cases, reflecting isolated ON as the initial manifestation of SLE rather than as a part of broader central nervous system (CNS) involvement [6]. Its rarity and potential early appearance have led some researchers to explore the utility of ON as an early diagnostic indicator of SLE [6].
ON is an ocular neuropathy with potential devastating consequences, including decreased visual acuity, orbital and ocular pain, metamorphopsias, and even blindness [10-12]. ON is commonly indicative of underlying autoimmune demyelinating disorders such as multiple sclerosis (MS), neuromyelitis optica spectrum disorder (NMOSD), and myelin-oligodendrocyte glycoprotein antibody-associated disease (MOGAD) [13]. In fact, about 50% of patients with ON go on to develop neurological symptoms suggestive of MS, and those with more severe ON often develop NMOSD [14-16]. It is important to note that SLE can occur concurrently alongside other inflammatory disorders [14,15]. ON can also be caused by both infectious and non-infectious inflammation [17]. These underlying causes result in occlusive vasculitis of the small arterioles of the optic nerve and edema of the myelinated nerve sheath with ensuing axonal necrosis and inflammatory demyelination of the optic nerve [9].
Given the prevalence of other underlying causes of ON, this systematic review will illustrate a diagnostic approach to (1) distinguish ON resulting from an autoimmune etiology as opposed to viral or other origins and (2) refine the diagnosis to suggest SLE, another autoimmune disorder, or concurrent conditions. Specifically, serological, radiological, and clinical features across SLE, MS, and NMOSD will be evaluated to guide differential diagnosis. This comprehensive approach incorporates factors, including antibody profiles, MRI findings, cerebrospinal fluid (CSF) studies, disease duration, and neurological symptoms. This study is unique in its holistic approach to provide a contemporary and inclusive overview of the defining characteristics of SLE, MS, and NMOSD, particularly in cases where ON is implicated.
Review
Methodology
Although this comprehensive literature review was not conducted in strict accordance with the Preferred Reporting Items for Systematic reviews and Meta-Analyses (PRISMA) guidelines, several key elements aligned with PRISMA standards and were incorporated to ensure methodological rigor. A structured and replicable search strategy, defined inclusion and exclusion criteria, and an independent assessment of study quality and risk of bias by multiple reviewers were utilized. The deviation from full PRISMA adherence was primarily due to the exploratory nature of the review and the specificity of its focus, which did not require formal registration or full adherence to reporting checklists typically associated with larger-scale systematic reviews. Still, details of the search process, study selection, and quality assessment are presented below to ensure transparency and reduce the potential for bias in the identification and analysis of relevant literature. Additionally, due to significant heterogeneity in study designs, populations, and outcome measures among the included studies, a quantitative synthesis such as a meta-analysis or meta-regression was deemed inappropriate. As such, a narrative synthesis was performed to qualitatively summarize and compare the diagnostic features across different etiologies of ON. This allowed for a more comprehensive descriptive analysis while acknowledging the methodological differences between the included studies.
A literature search for the identification of relevant studies was conducted on June 5, 2023, using the following electronic databases: PubMed (https://pubmed.ncbi.nlm.nih.gov/, accessed June 5, 2023) and Ovid Medline (accessed June 5, 2023).
The context of this review was to evaluate the current literature to compare the systemic and clinical diagnostic features between patients with SLE-associated ON and ON caused by other etiologies. The search strategy was to identify the most effective MeSH terms using the PubMed Search manager. This was done to determine if the MeSH terms produced articles that accurately fit the topic of the study. This was also done to find corresponding keyword equivalences to increase the sensitivity of the literature search. The MeSH terms included autoimmune disease, optic neuritis, multiple sclerosis, neuromyelitis optica, systemic lupus, ocular, idiopathic, diagnosis, complication, cause, features, and characteristics. Each of these was searched without date limitations. The search terms were then paired into the following: “autoimmune disease, optic neuritis, features,” “autoimmune disease optic neuritis features, multiple sclerosis,” “autoimmune disease optic neuritis features, neuromyelitis optica spectrum disorder,” “autoimmune disease optic neuritis features, systemic lupus erythematosus,” “underlying diagnosis and characteristics for optic neuritis,” “ocular conditions associated with systemic lupus erythematosus,” “systemic lupus erythematosus optic neuritis,” “ocular manifestation, systemic lupus erythematosus,” “association, optic neuritis,” “cause, optic neuritis,” “systemic lupus erythematosus, optic neuritis,” “optic neuritis, autoimmune disease,” “ocular complication, systemic lupus erythematosus,” “idiopathic disease, optic neuritis,” and “systemic lupus erythematosus, ocular.” None of these searches had any other filters applied to them, and all article types were included. The total number of articles identified was 2,954.
Approximately 10-20 articles from each search term were analyzed to determine if the article topics matched the topic of our review. After this process, “ocular manifestation, systemic lupus erythematosus,” “association, optic neuritis,” “cause, optic neuritis,” “systemic lupus erythematosus, optic neuritis,” “optic neuritis, autoimmune disease,” “ocular complication, systemic lupus erythematosus,” “idiopathic disease, optic neuritis,” and “systemic lupus erythematosus, ocular” were eliminated. Alongside these eliminations, duplicate articles were removed, leaving 2,621 articles. Further, “autoimmune disease optic neuritis features, multiple sclerosis” and “autoimmune disease optic neuritis features, neuromyelitis optica spectrum disorder” were eliminated due to their overlap with the search term “autoimmune disease, optic neuritis, features,” leaving 718 articles after putting the remaining topics into a PubMed search in a combined prompt with “OR” statements in between (PubMed automatically removed any duplicate publications at this stage).
The search terms were then coded into PubMed as follows: “(autoimmune disease, optic neuritis, features) AND ((autoimmune disease optic neuritis features, systemic lupus erythematosus) OR (underlying diagnosis and characteristics for optic neuritis) OR (ocular conditions associated with systemic lupus erythematosus) OR (systemic lupus erythematosus optic neuritis)).” This generated 28 articles. After applying the following filters: (1) publication dates from January 1, 2012, to May 5, 2023; (2) humans only; (3) English only; and (4) Medline articles (preprints excluded), 25 articles were identified. Further, after reading through each of the articles, one was eliminated due to lack of access, and another was eliminated for deviating from the topic (i.e., if their primary objective was not solely regarding diagnostic characteristics of one or more of the underlying causes of ON), resulting in 23 articles for analysis. The remaining articles were screened by examining the reference lists of all articles they included. No articles were eliminated in this process. Inter-reviewer agreement during the full-text screening phase was assessed using a consensus-based review. Although a formal Cohen’s kappa statistic was not calculated, discrepancies between the authors were resolved through discussion until agreement was achieved. A PRISMA-style flow diagram showing the article selection process is illustrated in Figure 1.
Flow diagram of the article selection process.
The data extracted from the selected publications included any information about epidemiology, symptoms, treatment protocols, and diagnostic factors for SLE, MS, and NMOSD, along with the considerable limitations of the study. During the review process, the quality and risk of bias for each included study were independently assessed by three authors using a customized checklist based on the National Institutes of Health (NIH) Study Quality Assessment Tools [18]. The checklist evaluated criteria such as clarity of research objectives, specification of study population, validity and reliability of outcome measures, and control of key confounding variables. While a standardized scoring rubric was not applied, each study was independently reviewed by the authors, and final assessments of risk of bias (categorized as low, moderate, or high) were determined through group consensus before proceeding with data synthesis.
Our study did not require ethical board approval because it solely reviewed previously published studies and did not involve human participants or identifiable patient information.
Results
In total, 23 articles, including eight retrospective studies, five cross-sectional studies, two cohort studies, four case reports, one narrative review, and three systematic literature reviews regarding the presence of ON in patients with SLE, MS, NMOSD, or a combination of the three, were analyzed. All studies were conducted within either outpatient or inpatient hospital settings. The study type, country of study origin, year of publication, and population size are presented in Table 1. Additional information regarding the main findings and limitations of each study can be found in the Appendices.
The assessment of the risk of bias for each included study was conducted as described in the Methodology section. Overall, studies with a low risk of bias were described as having robust methodologies, adequate sample sizes, clear definitions, and appropriate statistical analyses. Studies with a high risk of bias had significant methodological concerns, including very small to small sample sizes, lack of a control group, or unclear measurement techniques. Ultimately, reviewers categorized nine studies as having a high risk of bias, 12 studies as having a moderate risk of bias, and two studies as having a low risk of bias. Detailed explanations for each of the assessments can be found in the Appendices. Especially for the studies with a high risk of bias, greater caution was applied when integrating conclusions from them, whereas findings from studies with moderate to low risk were prioritized in identifying diagnostic patterns. This approach strengthened the credibility of our conclusions, allowing them to be grounded in the most methodologically sound evidence available.
Discussion
Studies Included
This review synthesizes information from a variety of studies, emphasizing a heterogeneous selection of study designs. This diversity, while providing a broader perspective, introduces a main limitation of this study. Variations in methodological rigor, sample size, and data reporting may have limited the ability to draw uniform conclusions across studies. These differences may also contribute to inconsistencies in the interpretation of diagnostic features.
Epidemiology
SLE and SLE with transverse myelitis (SLE-TM) primarily affect women between 15 and 44 years old, with female-to-male ratios of 9:1 and 8:1, respectively [19-22]. Ethnic minorities, especially those of African and Asian descent, have a higher prevalence of SLE at 27% [21]. The average age of onset differs slightly between SLE cases with and without CNS involvement. Disease durations for SLE cases with CNS involvement are reported to be 5.7 ± 8.2 years, while those without CNS involvement have an average duration of 9.2 ± 7.8 years [23]. Of note, regional variation and socioeconomic disparities may significantly impact access to diagnostic tools and long-term outcomes of SLE-associated ON [2-4]. Specifically, settings with lower socioeconomic status may have delayed diagnosis and limited access to advanced imaging or specialist care, resulting in worse neurological outcomes and higher rates of permanent visual impairment or blindness [2-4].
MS is more common in individuals aged 35 years or older, with a ratio of 66:34 for the ≥35-year to 20-34-year age groups. The female-to-male ratio for MS is 73:27, which is more balanced compared to SLE [20,24]. MS has a higher prevalence among individuals of European descent [20]. From 2003 to 2015, there was a significant increase in MS prevalence, while the incidence remained relatively consistent [24,25].
NMOSD is also more common in individuals aged 35 years or older, with a ratio of 73:27 for the ≥35-year to 20-34-year age groups [24]. Approximately 41.7% of NMOSD cases followed a previous diagnosis of SLE or primary Sjögren’s syndrome (pSS) [26]. The sex ratio for NMOSD has shown a significant change over time. From 2001 to 2005, it was slightly more common in females aged 35 years or older. However, between 2011 and 2015, it became vastly more common in females in the same age group. Overall, NMOSD appears more frequently in females, with a ratio of 78:22 and an average age of 45.1 ± 14.6 years [24,26]. The disease course tends to be worse in patients younger than 12 years old [19]. The incidence and prevalence of NMOSD have increased over time, with higher rates observed among certain ethnic minorities, particularly individuals of Mexican or Japanese origin [24,26,27].
Optic Neuritis Features
SLE-associated ON is rare, accounting for fewer than 1% of cases [20]. It typically presents bilaterally, with involvement of the optic chiasm, severe visual impairment, and intense pain exacerbated by eye movements [20,22]. Visual acuity recovery to better than 20/25 is experienced by only 50% of SLE-associated ON patients, compared to 87% of idiopathic ON and MS patients [20]. Some cases of SLE-associated ON manifest unilaterally as thrombotic optic neuropathy associated with antiphospholipid antibodies [20].
In NMOSD-associated ON, the condition usually manifests as single or recurrent episodes, including bilateral simultaneous ON or sequential ON occurring rapidly one after another [28]. Among patients with recurrent ON, a study observed that 12% developed NMOSD and 14% developed MS within five years of the initial episode [28]. The risk of developing MS continued to increase beyond the five-year mark, while the risk for NMOSD plateaued [28]. The coexistence of SLE and NMOSD has been frequently reported, and MRI evidence of ON, TM, and elevated aquaporin-4 (AQP4) antibodies is used to confirm the presence of NMOSD in SLE patients [29].
In MS-associated ON, the condition typically presents unilaterally with reduced visual acuity, diminished color vision, mild disc swelling, mild pain upon eye movement, and an afferent pupillary defect [20]. Improvement of symptoms should begin within three weeks of onset [20]. Rarely, MS-associated ON can manifest bilaterally, leading to severe vision loss, photophobia, vitritis, neuroretinitis, and other complications [20]. About 28% of MS-associated ON cases are recurrent [25]. The features of ON between these three conditions are summarized in Table 2.
Neurological Symptoms
Neurological symptoms can indicate various conditions, including autoimmune diseases, infections, neoplasms, vascular conditions, trauma, and nutritional deficiencies [22]. One-third of SLE cases primarily present with neuropsychiatric involvement, while severe CNS involvement is seen in 4.3% of cases [23,30]. Symptoms range from epileptic seizures, strokes, myelopathy, psychosis, to aseptic meningitis. Mild CNS involvement may manifest as mild cognitive dysfunction, headaches, depression, anxiety, and paresthesias [23]. Epileptic seizures in SLE patients are often accompanied by glomerulonephritis, lupus nephritis, and posterior reversible encephalopathy syndrome on MRI [23]. Demyelination is almost always present in neuropsychiatric SLE and shares similar patterns with NMOSD [30].
Transverse myelitis (TM) is rare in SLE patients, occurring in 1-2% of cases [18,21]. It can manifest within the first five years after SLE diagnosis, often as a monophasic illness with poor recovery [19,22]. However, highly recurrent SLE-related longitudinally extensive transverse myelitis (LETM), a disease of contiguous, inflammatory spinal lesions that cause symptoms of paralysis, paresthesias, and urinary retention, is also reported [22]. NMOSD is characterized by ON and LETM, which are commonly present alongside acute brainstem syndrome and its symptoms of diplopia, intractable hiccup, nausea, and vomiting [22,26,30]. LETM in NMOSD typically manifests bilaterally with weakness and sensory disturbances, and there can be intervals between attacks as well [22,26]. Motor and sensory symptoms, loss of sphincter control, and tonic spasms can occur [26]. Gastrointestinal autoimmune diseases are commonly associated with MS but not with NMOSD [27].
In MS, immune cells infiltrate across the blood-brain barrier, promoting inflammation, demyelination, gliosis, and neuroaxonal degeneration of the CNS’s white matter, as apparent in the inflammatory demyelination often found in brain biopsies of MS patients [15]. This may result in asymmetrical, progressive myelitis, and short-segment spinal lesions in less than half of the spinal cord diameter, and may include nodular enhancements [15]. Further, cortical lesions are common, while thalamic/hypothalamic lesions are uncommon [15]. CSF analysis shows oligoclonal bands in >90% of patients, although CSF is usually normal [14]. In one study, myasthenia gravis has been reported to be slightly associated with MS at a rate of 0.9% (24/4,627 patients) [24].
Antibody Profiles
The primary biomarker indicating NMOSD is the presence of anti-aquaporin-4 (AQP4) IgG antibodies, but these may be absent in 5-40% of NMOSD patients [22,26,30-33]. As TM can be an initial symptom in both NMOSD and MS, examining serum AQP4-IgG levels in TM patients can aid in distinguishing between NMOSD and MS. NMOSD can coexist with other autoimmune disorders such as SLE, myasthenia gravis, and antiphospholipid syndrome [29,30,32]. In SLE patients who later develop NMOSD, 79% were found to have AQP4-IgG antibodies [30]. Another distinguishing marker is the presence of neuromyelitis optica immunoglobulin (NMO-IgG), which binds to AQP4 channels and indicates high-risk syndromes [22,28,34]. However, NMO-IgG antibodies are not consistently detected in NMOSD patients, although their presence predicts disease relapse [22,28,31,34]. In cases of LETM, the presence of AQP4-IgG antibodies is useful for distinguishing NMOSD (positive) from MS (negative) [22,32]. It is crucial to recognize that other antibodies, such as myelin-oligodendrocyte glycoprotein-IgG, may also be involved in NMOSD pathophysiology, especially in AQP4-IgG-negative patients [30]. Another factor in ruling out MS is the presence of acetylcholine receptor antibodies, found in 11% of NMOSD patients but not in MS or healthy individuals [29].
In SLE, the typical antibody profile includes antinuclear antibodies (ANAs), anti-single-stranded DNA (anti-ssDNA), anti-double-stranded DNA (anti-dsDNA), lupus anticoagulant (LAC), anticardiolipin antibodies (aCL), and anti-Sjögren’s syndrome-related antigen A/B (anti-SSA/SSB) antibodies [26,27,31,32]. It is important to note that these antibodies may be absent in some patients with clinically evident SLE, especially when the disease is in its early phases or presents atypically [26,32]. The absence of specific autoantibodies can complicate diagnosis and delay treatment, particularly in patients who present with neurologic symptoms suggesting the presence of overlapping autoimmune syndromes [26,32]. Generally, the presence of AQP4 antibodies is associated with seropositivity for SSA, SSB, ANA, and dsDNA antibodies [33]. The overlapping serological profiles of SLE, MS, and NMOSD underscore the diagnostic challenge of distinguishing a single one of these autoimmune disorders with the presence of autoantibodies alone. Additionally, MS patients may test positive for anti-Smith (Sm), ssDNA, and LAC antibodies, while NMOSD patients may be seropositive for aCL antibodies [27]. The interrelationships in autoantibody expression increase the risk of missclassification between demyelinating diseases and systemic autoimmune conditions. The presence of non-specific autoantibodies in MS or NMOSD, for instance, can mimic SLE-related neuroinflammation, requiring a careful and comprehensive diagnostic approach to avoid inappropriate management of these patients.
Imaging Findings
When suspecting a systemic disorder, a diagnostic approach typically involves cranial MRI scans, although full-body positron emission tomography scans are more suitable when there is suspicion of neoplasms [35]. The diagnostic criteria for differentiating between MS and NMOSD involve MRI. MRI also plays an integral role in distinguishing them from other neurological conditions [35]. The utilization of ultra-high-field strength (7-T) MRI, coupled with innovative 7-T T2*-weighted fast low-angle shot imaging, enables the identification of brain and spinal cord lesions, recognition of central vein signs, and detection of leptomeningeal enhancement. These advanced imaging techniques increase the sensitivity and specificity for diagnosing both MS and NMOSD [22,26,35,36].
In NMOSD, substantial and extensive plaques are associated with high AQP4 expression, primarily located in the periventricular and hypothalamic areas [26,31,34,36]. These plaques exhibit colocalized blood vessels in their periphery [31,36]. However, unspecified white matter lesions (WMLs) are also highly common in NMOSD [36]. Surprisingly, T2*-weighted images show a lack of hypointense rimming and, on rare occasions, the presence of crossing vasculature, despite the prominent vascular involvement in NMOSD [36].
In contrast to NMOSD lesions, MS plaques predominantly surround a small vein and exhibit hypointense rimming, suggesting neuroinflammation from vascular or unspecified WMLs [36]. T2*-weighted imaging reveals that 80% of MS lesions, with 19% originating from asymptomatic WMLs, are perivascular, indicating their predictive value for demyelinating diseases [36]. Furthermore, perivascular lesions, present in 92% of MS patients using 7-T MRI, are a requirement for classifying MS, as they demonstrate the presence of a vein within cerebral plaques [36]. Cortical lesions are also highly prevalent in MS [36]. The absence of MS-like lesions in asymptomatic patients confirms the unique morphology of MS plaques. Patients with SLE, on the other hand, have lesions spanning more than four vertebral bodies in approximately 60% of the cases [22].
Dermatological Symptoms
Pruritus, a neural pathway-induced sensation, has been identified as an early sign of NMOSD, often preceding weakness [32]. In one study, pruritus was observed in 28% of NMOSD patients, all of whom had NMO-IgG in their serum [34]. This sensation did not always align with the dermatomal distributions of the affected spinal cord segments, implying a spinal cord lesion. The precise mechanism underlying this sensation remains unclear. However, rodent studies suggest that trauma, tumors, and inflammation stimulate afferent neurons in the dorsal horn [34].
NMOSD also presents cutaneous and joint findings, with Raynaud’s phenomenon being the most common cutaneous manifestation [37]. Conversely, SLE commonly displays widespread dermatological manifestations, including malar rash, discoid rash, photosensitivity, and non-scarring alopecia, all of which are included in the current diagnostic criteria for SLE [32].
Genetic Involvement
The general transcription factor II-I (GTF2I) gene encodes transcription factor II-I (TFII-I), described primarily in mice as a crucial regulator of T and B-cell activation through vascular endothelial growth factor receptor 2 (VEGFR2) gene deregulation. In humans, its role has been characterized mainly in the regulation of other genes [38]. A robust genome-wide association study involving six East Asian cohorts confirmed a correlation between the GTF2I rs733666469 polymorphism and SLE occurrence. Another study suggested a potential association between the T allele of the GTF2I rs117026326 C/T polymorphism and SLE occurrence. However, further research is needed to establish the significance of this latter polymorphism [38]. Although the mechanism is not yet fully understood, the GTF2I rs117026326 polymorphism is also linked to NMOSD and MS [38]. Moreover, GTF2I polymorphisms, in general, have been found to be associated with rheumatoid arthritis, primary Sjögren’s syndrome, and other pathologies in Asian cohorts [38].
Bloodwork
Serum semaphorin 3A (SemaA3), recognized as an immunoregulatory molecule involved in oligodendrocyte regeneration and suppression of T cells, and its receptor neuropilin-1 have been implicated in the pathogenesis of SLE and other autoimmune diseases. Their levels are inversely correlated with SLE disease activity [30]. SLE patients are also prone to developing leukocytopenia and reduced complement levels [19,32]. Similarly, NMOSD patients may exhibit low serum complement levels [32].
Compared to healthy individuals, both MS and NMOSD patients have significantly elevated levels of low-density granulocytes (LDGs). However, the occurrence of LDGs in MS and NMOSD is not as frequent as in SLE patients [39]. In the study reported by Ossendorf et al., of the 23 healthy donors, none reached the 0.7% threshold (0.2%), while a majority of the 17 (0.9%) MS and 20 (2.1%) NMOSD patients did [39]. Nonetheless, the levels of LDGs in both MS and NMOSD do not match those found in SLE (4.3%) [39]. Although the exact mechanism remains unclear, the prevailing thought is that LDGs are a byproduct of ongoing inflammation rather than a part of the disease pathogenesis [39-41].
Other Findings
SLE-TM patients are more likely to experience fever compared to patients with SLE alone. The systemic inflammation caused by TM in SLE contributes to the higher incidence of fever [19]. A CSF white blood cell count exceeding 50 cells/µL indicates a possible infectious etiology, necessitating subsequent polymerase chain reaction analysis of CSF and additional viral serologies [22].
Conclusions
SLE-associated ON, though rare, has diagnostic value as an early indicator of CNS involvement. Unlike ON associated with MS or NMOSD, SLE-associated ON tends to be bilateral, more severe, and less responsive to treatment. Through this systematic review, it is evident that no single clinical feature or diagnostic test is sufficient alone. Instead, a multidisciplinary approach, incorporating neuroimaging, serological markers such as AQP4-IgG, and patient demographics, provides the most robust framework for distinguishing among these autoimmune diseases. As a result, clinicians will be able to make more timely and precise diagnoses. Early recognition of ON in SLE not only facilitates prompt immunosuppressive treatment but may also alter the long-term neurological trajectory of patients. Additionally, emerging biomarkers, including LDGs and semaphorins, have shown early promise in identifying disease activity and may offer novel pathways for early diagnosis and monitoring in SLE-associated ON. Future research should aim to develop unified diagnostic criteria and explore novel biomarkers specific to SLE-associated ON. Further investigation of key biomarkers, as well as genetic and inflammatory mediators, could enhance early detection and lead to improved therapeutic strategies. Ultimately, integrating ocular symptoms such as ON into the diagnostic paradigm of SLE promises to enhance early detection, reduce morbidity, and improve quality of life for patients navigating this complex and often debilitating disease.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Systemic lupus erythematosus: new diagnostic and therapeutic approaches Annu Rev Med Lazar S Kahlenberg JM 339352742023 https://pubmed.ncbi.nlm.nih.gov/35804480/3580448010.1146/annurev-med-043021-032611 · doi ↗ · pubmed ↗
- 2Rising incidence and prevalence of systemic lupus erythematosus: a population-based study over four decades Ann Rheum Dis Duarte-García A Hocaoglu M Valenzuela-Almada M 12601266812022 https://www.cdc.gov/lupus/data-research/index.html 3557738510.1136/annrheumdis-2022-222276 PMC 10481386 · doi ↗ · pubmed ↗
- 3Global epidemiology of systemic lupus erythematosus: a comprehensive systematic analysis and modelling study Ann Rheum Dis Tian J Zhang D Yao X Huang Y Lu Q 351356822023 https://pubmed.ncbi.nlm.nih.gov/36241363/3624136310.1136/ard-2022-223035 PMC 9933169 · doi ↗ · pubmed ↗
- 4Delayed diagnosis adversely affects outcome in systemic lupus erythematosus: cross sectional analysis of the Lu La cohort Lupus Kernder A Richter JG Fischer-Betz R 431438302021 https://pmc.ncbi.nlm.nih.gov/articles/PMC 7933718/3340203610.1177/0961203320983445 PMC 7933718 · doi ↗ · pubmed ↗
- 5Systemic lupus erythematosus in primary care: an update and practical messages for the general practitioner Front Med (Lausanne) Gergianaki I Bertsias G 16152018 https://pubmed.ncbi.nlm.nih.gov/29896474/2989647410.3389/fmed.2018.00161 PMC 5986957 · doi ↗ · pubmed ↗
- 6Optic nerve involvement in childhood onset systemic lupus erythematosus: three cases and a review of the literature Lupus Suri D Abujam B Gupta A 9396252016 https://journals.sagepub.com/doi/abs/10.1177/09612033156031422634124310.1177/0961203315603142 · doi ↗ · pubmed ↗
- 7Systemic lupus erythematosus presenting as optic neuropathy: a case report Cureus Zahid S Iqbal M 0112019 https://pmc.ncbi.nlm.nih.gov/articles/PMC 6682382/10.7759/cureus.4806 PMC 668238231403006 · doi ↗ · pubmed ↗
- 8Optic disc swelling as the first presentation of systemic lupus erythematosus: a case report Egyptian J Hosp Med Abdulqader A 633637802020 https://www.ejhm.journals.ekb.eg/article_92542.html