Molecular analysis of T-cell Acute Lymphoblastic Leukemia arising after Essential Thrombocythemia foreshadows a distinct clonal route for lymphoid blast crisis in Philadelphia-negative chronic myeloproliferative neoplasm: a case report with literary review
Francesco Grimaldi, Mara Memoli, Simona Avilia, Roberta Russo, Giulia Scalia, Roberta Visconti, Santa Errichiello, Barbara Izzo, Fabrizio Pane

TL;DR
This case study explores a rare progression from Essential Thrombocythemia to T-cell Acute Lymphoblastic Leukemia, suggesting a new clonal pathway for lymphoid blast crisis in chronic myeloproliferative neoplasms.
Contribution
The study provides molecular evidence supporting a distinct clonal origin for secondary Acute Lymphoblastic Leukemia in Philadelphia-negative chronic myeloproliferative neoplasms.
Findings
Molecular analysis supports a different clonal route for lymphoid blast crisis in Philadelphia-negative chronic myeloproliferative neoplasms.
Secondary Acute Lymphoblastic Leukemia should be viewed as a separate cancer rather than a true blast crisis.
A review of published cases highlights the rarity and unique features of MPN transforming to Acute Lymphoblastic Leukemia.
Abstract
Progression to Acute Myeloid Leukemia is a well-known complication of classical philadelphia-negative chronic myeloproliferative neoplasms, while disease progression to Acute Lymphoblastic Leukemia remains an extremely unfrequent event. A molecular explanation for this rare phenomenon is missing. However, the clonal haematopoiesis mostly present in these patients may work as a seeding soil for a second neoplastic disease. Molecular results from this case study reporting a secondary Acute Lymphoblastic Leukemia presenting after Essential Thrombocythemia support this hypothesis. In this contest secondary Acute Lymphoblastic Leukemia should not be considered as real blast crisis, but rather as a second cancer determined by a different clonal route. Given the unique features of this case, a review of the published cases of MPN transforming to Acute Lymphoblastic Leukemia available in…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
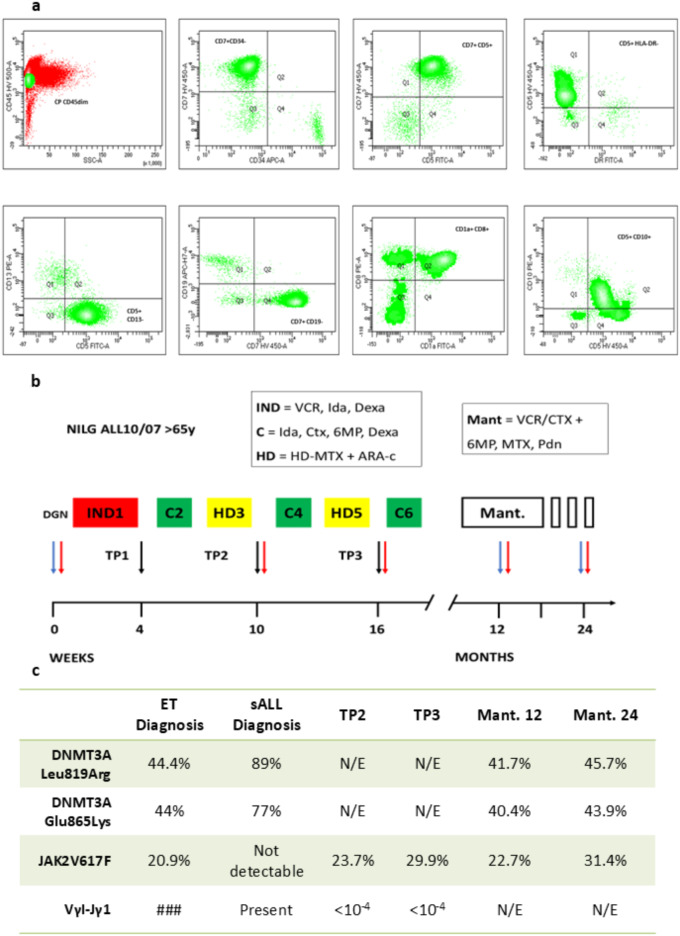 Figure 1
Figure 1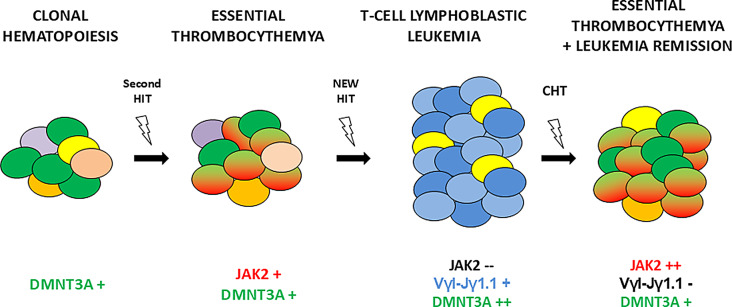 Figure 2
Figure 2- —Università degli Studi di Napoli Federico II
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsMyeloproliferative Neoplasms: Diagnosis and Treatment · Acute Myeloid Leukemia Research · Chronic Myeloid Leukemia Treatments
Introduction
Progression to Acute Myeloid Leukemia (AML) is a well-known complication of classic Philadelphia-negative chronic myeloproliferative neoplasms (MPN) including Polycythaemia Vera (PV), Essential Thrombocythemia (ET) and Primary Myelofibrosis (PMF) [1]. The incidence of leukemic transformation varies across MPN subtypes, reaching 10–20% at 10 years in PMF, followed by PV, where incidence is reported about 2.3% at 10 years and 7.9% at 20 years, and finally in ET, where transformation risk is expected to be less than 3% at 10 years, predominantly in cases later reclassified as prefibrotic PMF [2–4]. Currently, JAK2, CALR and MPL mutations are considered to play a pivotal role as driver events in MPN pathogenesis [5], while leukemic evolution is commonly associated with accumulation of additional mutations in different myeloid cancer genes, such as RUNX1, ASXL1, IDH2, and TP53, that contribute to increase clonal complexity and heterogeneity in the hematopoietic stem cell (HSC) compartment [6, 7].
In contrast, transformation to secondary Acute Lymphoblastic Leukemia (sALL) is unusual and exceedingly rare in MPNs [8]. Due to paucity of molecular and clinical data available [9, 10], mechanism of lineage switch from a chronic MPN to a lymphoid blast crisis remains elusive. Given that most of reported cases have been associated with B-lineage ALL and are associated with JAK2 positivity [9, 10], and that JAK2 mutation has been detected in B/NK/T-lymphoid precursors from PV and PMF patients [11, 12], it has been speculated that the aberrant JAK2 tyrosine kinase activity in a common myelo-lymphoid CD34 + progenitor may play a causative role and contribute to lymphoid transformation. Nevertheless, some studies [13, 14] have described sALL cases harboring a molecular profile completely distinct from the antecedent MPN, implying independent clonal origins.
In according with these observations, we report the case of a patient with JAK2-V617F–positive ET who developed more than a decade later a T-cell sALL. Molecular analyses performed during the disease course clearly revealed a JAK2-negative lymphoid clone arising from a persistently DNMT3A-mutated clonal background, supporting a model in which clonal hematopoiesis (CH) may contribute to a dual neoplastic evolution, with a different clonal route for the rare sALL diagnosed after MPNs.
Case description
In October 2012 a 62 year-old women was initially referred to our clinic for thrombocytosis. JAK2-V617F molecular test (Supplementary methods, S1) was positive on peripheral blood and a bone marrow biopsy confirmed the histological diagnosis of ET.
Patient was started on hydroxyurea, and continued on with good response and without thrombotic events over the following ten years. A decade later, she was urgently admitted with high fever, fatigue and joint pain. Complete blood counts at time showed: Hgb 10,4 g/dl, WBC 20.680/mm3, PLT 23.000/mm3. Peripheral blood and bone marrow smear revealed lymphoid blast cells (80% and 95% respectively), with following surface markers at flow-cytometry analysis: CD45dim, DR-, CD34-, CD5+, CD7+, CD2+, CD8+, CD1a+, CD99+, CD10+, CD3-, cyCD3+/- (Supplementary methods, S3). According to EGIL classification, diagnosis of secondary Cortical T-ALL was made (Fig. 1a). Standard cytogenetic, with FISH study to detect chromosome 9, 11 and 17 aberrations, revealed a normal karyotype (46 XX), while molecular testing by RT-PCR for SIL/TAL, NUP98 rearrangments and TAF1/NUP214 was negative. Remarkably, the JAK2-V617F mutation became undetectable in the bone marrow at diagnosis of sALL. With RT-PCR, a disease specific VγI-Jγ1.1 IGH sequence was identified for monitoring of minimal residual disease (MRD) (Supplementary methods, S1). Patient started induction chemotherapy according to NILG 10/07 ALL protocol [15] for elderly patients (Age > 65 years). Bone marrow evaluation performed after cycle 1, 3 and 5 of chemotherapy (Time point 1, 2, 3; see Fig. 1b) showed complete remission (CR), with molecular MRD constantly negative (below < 1 × 10-4 level of test sensitivity). Interestingly, JAK2-V617F mutation became again detectable in the bone marrow, with an increase in allele burden of 23,7% at Tp2 and 29,9% at Tp3. Patient successfully completed the planned 6 cycles of chemotherapy and started maintenance therapy. Bone marrow evaluation performed after first and second year of maintenance confirmed the CR status, with no blast cells detected by flow-cytometry, and JAK2-V617F positivity with a stable allele burden of 22,7% and 31.4% respectively. To further investigate the clonal origin of the T-cell ALL, a Next-Generation Sequencing (NGS) analysis using a 36-gene myeloid panel (Supplementary methods, S2) was performed on bone marrow samples collected and previously stored at four time points: ET diagnosis, sALL diagnosis, and during first- and second-year maintenance. NGS analysis revealed two missense mutations in DNMT3A gene, occurring on exon 21 (Leu819Arg; T2456 > G) and exon 22 (Glu865Lys; G2593 > A), known to be pathogenic and related with clonal haematopoiesis (CH). Interestingly, these two mutations were already detectable at the time of ET diagnosis, with a variant allele frequency (VAF) of 44.4% (for exon 21 mutation) and 44% (for exon 22 mutation). Their VAFs increased to 89% and 77% at the time of T-ALL diagnosis, respectively. Despite intensive chemotherapy, the VAFs returned to relatively stable levels of 41.7% (exon 21) and 40.4% (exon 22) after the first year of maintenance, and of 45.7% (exon 21) and 43.9% (exon 22) after the second year (see Fig. 1c). Finally, to assess penetrance of DNMT3A and JAK2 mutations in pre-leukemic HSC compartment, NGS analysis was performed on sorted B and T-lymphocytes in peripheral blood, even if only from peripheral blood collected at second year of maintenance (Supplementary methods, S2, S3). Very interestingly, no JAK2 mutations were detected in either lineage. However, both DNMT3A mutations were identified in T-cells, even if with a low allele frequency below limits of assay detection (VAF below 5%), suggesting its persistence in early hematopoietic progenitors (Supplementary Methods, S2–S3).
Fig. 1a: Diagnosis of secondary T-ALL (sALL). b: NILG-ALL 10/07 regimen. IND = induction, C = consolidation, HD = high dose chemotherapy, Mant = maintenance. VCR = vincristine, IDA = idarubicin, Dexa = Dexamethasone, Ctx = cyclophosphamide, 6MP = 6-mercaptopurin, MTX = Methotrexathe, PDN = prednisone. Arrows = bone marrow sampling; Black arrow = MRD sampling, Red arrow = JAK2 allelic burden, Blue arrow = NGS sampling. c: mutations kinetics observed from ET diagnosis trough sALL treatment
Discussion with literature review
Several studies have reported the incidence of secondary acute lymphoblastic leukemia (sALL) following a prior malignancy. A large SEER analysis [16] found that 6.6% of newly diagnosed ALL cases in the United States occurred as secondary malignancies. A retrospective series from the GIMEMA group reported a lower incidence of 2.3% [17], while Aldoss et al. [18] more recently described an incidence of 9.3% at a single U.S. institution. In all these cohorts, fewer than 10% of sALL cases followed a primary hematologic malignancy, and transformation from MPN to sALL remains a rare and anecdotal event. Given the rarity of this case, we conducted a literature review of published cases of MPN transforming to ALL. We searched PubMed using the following keywords: “acute lymphoblastic leukemia, myeloproliferative neoplasm, chronic myeloproliferative, essential thrombocythemia, polycythemia vera, myelofibrosis”. After removing reviews and other irrelevant papers, we identified a total of 31 patients reported in literature from 1978 to the present, including the current case. The complete patient series is reported in Table 1 with references.
Across these 31 identified patients, median time to leukemic transformation from initial diagnosis of MPN was of 10 years. In 15 patients a molecular analysis of MPN phase was available, with a JAK2V617F mutation identified in 66% of cases (10/15). The major part of patients (78%) showed a B-cell phenotype after leukemic transformation (24/31), and, despite intensive chemotherapy, only 25% of patients resulted alive at reported follow-up. Prevalence of sALL incidence in JAK2-mutated MPN have stimulated the idea that a common JAK2-mutated myelo-lymphoid precursor could explain the origin of this condition, sustained by the experimental evidence that generally a low JAK2 allelic burden (3–5%) can be found both in B- and T-lymphocytes of MPN patients [10, 11].
However, in our case JAK2-V617F canonical mutation disappeared at the sALL onset, and became again detectable after the patient obtained the CR, with an allele burden progressively increased during maintenance therapy. The disappearance of JAK2V617F at in the moment of diagnosis, when the blast size in bone marrow is relevant, have been already described even with differently mutated MPN (CALR-mut or MPL-mut; see Table 1) and strongly suggests that the leukemic transformation may occur independently of the original MPN-defining mutation. These findings finally align with previous molecular case studies indicating distinct clonal origins for MPN and sALL phases [13, 14].
Table 1. Summary of cases of MPN transformed into sALL reported in literatureCase no.YearAge / genderMPNsubtypeMolecular statusMPNtreatmentTime to progression (years)s-ALL PhenotypeCytogenetics(of sALL)Clinical outcomeReference1197877 / MPVNAPhlebotomy, CHL6B-cellNormalDead [19]2198061 / MPMFNAOxymetholone, splenectomy5B-cellAneuploidDead [20]3198053 / FPPV-MFNAPhlebotomy18Burkitt’sNADead [20]4198674 / MPVNAP32, Bu, CHL, BCNU6NullNADead [21]5198742 / MPVNAP32, Bu10NullDel 6q, +8Dead [22]6198720 / MPVNAP32, Bu10T-cellNRDead [22]7198868 / FPVNAP32, Bu, CHL25B-cellComplexDead [13]8199354 / FPVNAP3211B-cellt(9;22)Dead [23]9199476 / MPVNABU16B-cellDiploidDead [24]10199550 / METNAHU3B-cellComplexAlive [25]11199654 / FPVNAP32, HU13B-cellNAAlive [26]12199687 / FETNAHU1T-cellNANA [27]13199670 / FETNAP32, HU19B-cellDiploidNA [28]14199963 / MPPV-MFNAP32, HU6B-cellNADead [29]15200553 / MPMFNAHU2B-cellNADead [30]16201256 / MPMFNegHU1B-cellt(9;22), del 20qDead [31]17201254 / MPPV-MFExon 12Phlebotomy4B-cellDiploidDead [32]1820145 / FPVNAPhlebotomy17B-cellNANA [33]19201467 / FETV617FHU16B-cellt(9;22)Dead [14]20201565/ METV617FNA16B-cellDel (9) (p13)NA [34]21201559 / MET-MFV617FNA10B-cellDel 13q and 20qNA [34]22201572 / MPVV617FHU3B-cellNormalAlive [35]23201664 / FPVV617FHU4B-cellNormalAlive [36]24201665 / FETCALRHU3.5B-cellHyperdiploidAlive [37]25201769 / FETCALRHU, anagrelide, ruxolitinib26B-cellt(1;19)Dead [38]26201766 / FPMFV617FHU, lena11B-cellComplex monosomalDead [39]27201876 / MPPV-MFV617FPhlebotomy,ruxolitinib7B-cellNANA [40]28202062 / MPMFV617FEpo3T-celli(17)Dead [41]29202149 / METMPLHU10T-cellComplexAlive [42]30202167 / FETMPLHU13B-cellt(9;22)NA [43]31202277 / FETTriple-negHU2B-cellNAAlive [44]32202462 / FETV617F*HU10T-cellNormalAliveCurrent CaseET, essential thrombocythemia; PV, policythemia vera; PMF, primary myelofibrosis; PPV-MF, post-PV myelofibrosis; PET-MF, post-ET myelofibrosis; NA, not available in the report; HU, hydroxyurea; CHL, clorambucil; P32, Phosphorus-32; Bu, busulphan; *Mutation not detectable, or with VAF ≤ 5%, at leukemic transformation
JAK2-driven hemopoiesis re-expanded once the patient achieved CR, showing the ability of JAK2-mutated HSC to survive under stress conditions as ones given by chemotherapy, and as highlighted in several MPN disease models [45, 46]. NGS analysis identified two DNMT3A gene mutations that were present at the time of diagnosis of ET, increased in VAF only at the diagnosis of T-cell sALL, and then remained relatively stable during the entire course of the patient monitoring. Several studies of tumoral DNA from T-lymphoproliferative neoplasm [47, 48], and recently even from elderly patients with T-cell ALL [49, 50], have clearly identified an increased incidence of myeloid gene mutations related to CH, such as DNMT3A and TET2, in leukemic cells. In particular, Bond et al. [50], have analyzed implications of DNMT3A mutations in T-cell ALL, performing a comprehensive genetic and clinico-biological analyses of T-ALL treated during the GRAALL-2003 and − 2005 studies. They were able to show that, at least in two patients, DNMT3A mutations were present in non-leukemic cell DNA, providing the first potential evidence of age-related CH in T-cell acute lymphoblastic leukemia, and that DNMT3A mutation was associated with older age. In addition to this, very recently preliminary results from Horns et al. [51] showed that CH mutations are detectable in secondary B-cell ALL, and define a particular group of sALL diagnosed in patients exposed to lenalidomide for Multiple Myeloma treatment. Also the mutational profile of these patients showed a strong enrichment of CH genes in ALL, with three mutually exclusive patterns: TP53 mutated, IDH2 mutated and DNMT3A (Including non-R882) mutated. Altogether these data suggest that DNMT3A driven CH may contribute to ALL development, at least in older adults or in secondary ALL.
In according with these finding, in our patient the DNMT3A mutations were present at higher VAF (40%) than the JAK2-V617F mutation (20,9%) at the time of ET diagnosis, suggesting that the DNMT3A mutation occurred as first in the HSC compartment. This condition is concordant with experimental observations that determined the mutational hierarchy in which the HSC can acquire canonical JAK2 mutation [52]. In some cases, mutations in epigenetic regulators such as TET2 can be acquired before JAK2, and in these conditions the HSC compartment is completely dominated by TET2 single-mutant cells (TET2-first). NGS data from our patient are consistent with the TET2-first model of MPN (even if in our case the mutations involved a different epigenetic regulator as DNMT3A) and suggest that the HSC compartment was dominated by DNMT3A at the time of diagnosis of ET. Two other elements support this idea. Firstly, DNMT3A mutations has been detected at a very low level even in mature T-lymphocytes in our sorting experiment, suggesting its persistence in a hierarchically higher progenitor of HSC. Secondly, the DNMT3A mutation remained detectable in bone marrow during CR, similarly to what happens in AML patients, confirming their nature of CH mutations [53, 54].
Collectively, these data suggest that in our patient, an early DNMT3A-driven CH event created a clonal foundation that first facilitated the development of ET via JAK2 acquisition, and later, after an uncharacterized second genetic hit, led to the emergence of T-ALL (Fig. 2). However, once chemotherapy cleared all the blast cells in the bone marrow, the DNMT3A/JAK2 clone re-emerged and expanded under hematopoietic stress conditions. These findings support a model in which lymphoid blast crisis in MPNs arises from a distinct clonal trajectory independent of JAK2, CALR, or MPL mutations, yet rooted in a common CH background, as has been noted in other conditions [55, 56].
Fig. 2ET originates from clonal haematopoiesis when JAK2V617F mutation occurred on DNMT3a mutated cells. Years after, a new genetic hit selects a s-ALL clone and drives leukemia expansion over JAK2-mutated cells. After chemotherapy, under stress conditions, DNMT3a + JAK2 + clone re-expanded over polyclonal haematopoiesis, with an increasing allele burden
Conclusions
Philadelphia chromosome-negative MPNs are characterized by the presence of driver mutations (JAK2, CALR, and MPL), which play a central role in disease classification and pathogenesis. However, the importance of screening for additional somatic mutations in genes involved in epigenetic regulation (i.e. ASXL1, TET2, DNMT3A), RNA splicing (SRSF2, U2AF1, SF3B1), and signaling pathways (NRAS, KRAS, SH2B3) has become more relevant, considering their association with disease progression, leukemic transformation, and overall survival. Incorporating comprehensive molecular profiling into routine clinical evaluation enhanced in recent years diagnostic accuracy, risk stratification, and personalized treatment strategies with refined prognostic models such as MIPSS70 + and GIPSS [57]. Several of these mutations also occur in the context of CH, condition characterized by age-related acquisition of somatic mutations in HSC in the absence of overt hematologic malignancy. The overlap between CH-associated mutations and those found in MPNs suggests a continuum of clonal evolution, with early mutations potentially influencing MPN development and subsequent disease trajectory. In this view, the clonal dynamics tracked down from our study shed some lights on the mechanism of lymphoid blast crisis diagnosed in the contest of Philadelphia chromosome-negative MPNs. The disappearance of JAK2 mutation during sALL, followed by its re-emergence post-therapy, and the persistence of DMT3A mutation, strongly support the hypothesis of divergent clonal evolution from a common CH background. In this contest sALL observed after Philadelphia-negative MPN should not be considered as a real blast crisis, but rather as a second neoplasm originating from a shared clonal origin. Despite the rarity of these cases, additional genomic studies, possibly at a single-cell-level, will be needed to clearly understand and finally assess the origin of this rare phaenomenon, refining our understanding of clonal architecture in MPNs evolution.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplementary Material 1
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Annotation ID="1" Type="" Text="The following mandatory elements (Page numbers) of the reference are missing in the Manuscript. Please check and verify." Category="Completeness" Iurlo A, Cattaneo D, Gianelli U (2019) Blast transformation in myeloproliferative neoplasms: risk factors, biological findings, and targeted therapeutic options. Int J Mol Sci 20(8)10.3390/ijms 20081839 PMC 651480431013941 · doi ↗ · pubmed ↗
- 2Annotation ID="2" Type="" Text="The following mandatory elements (Page numbers) of the reference are missing in the Manuscript. Please check and verify." Category="Completeness" Wu D, Ye B, Shen J, Peng L et al (2016) Acute lymphoblastic leukemia in the course of polycythemia Vera: A case report and review of literature. Indian J Hematol Blood Transfus 3210.1007/s 12288-015-0598-y PMC 492552927408354 · doi ↗ · pubmed ↗
- 3Pagano L et al (1999) Acute lymphoblastic leukaemia occurring as second malignancy: report of the GIMEMA archive of adult acute leukaemia. Gruppo Italiano malattie ematologiche Maligne Dell’Adulto. Br J Haematol 106(4). 10.1046/j.1365-2141.1999.01636.x 10.1046/j.1365-2141.1999.01636.x 10520009 · doi ↗ · pubmed ↗
- 4Aldoss I, Stiller T, Tsai NC (2018) Therapy-related acute lymphoblastic leukemia has distinct clinical and cytogenetic features compared to de novo acute lymphoblastic leukemia, but outcomes are comparable in transplanted patients. Haematologica 103(10):1662–166810.3324/haematol.2018.193599 PMC 616579429903756 · doi ↗ · pubmed ↗
- 5Annotation ID="5" Type="" Text="The following mandatory elements (Year) of the reference are missing in the Manuscript. Please check and verify." Category="Completeness" Braich TA, Grogan TM, Hicks MJ et al Terminal lymphoblastic transformation in polycythemia vera. 1986 Am J Med 80:304–30610.1016/0002-9343(86)90029-x 3456200 · doi ↗ · pubmed ↗
- 6Annotation ID="6" Type="" Text="The following mandatory elements (Year) of the reference are missing in the Manuscript. Please check and verify." Category="Completeness" Neilson JR, Patton WN, Williams MD et al (1994) Polycythaemia rubra vera transforming to acute lymphoblastic leukaemia with a common immunophenotype. J Clin Pathol 47:471–47210.1136/jcp.47.5.471PMC 5020328027406 · doi ↗ · pubmed ↗
- 7Annotation ID="7" Type="" Text="The following mandatory elements (Year) of the reference are missing in the Manuscript. Please check and verify." Category="Completeness" Camos M, Cervantes F, Montoto S et al (1999) Acute lymphoid leukemia following polycythemia vera. Leuk Lymphoma 32:395–39810.3109/1042819990916740410037041 · doi ↗ · pubmed ↗
- 8Gawel WB, Helbig G, Boral K, Kyrcz-Krzemien S(2016) Acute lymphoblastic leukemia transformation in polycythemia vera: a rare phenomenon. Indian J Hematol Blood Transfus 32(open in a new window):62–65. Epub 2016/ 07/1410.1007/s 12288-016-0673-z PMC 492556327408357 · doi ↗ · pubmed ↗