Navigating the neuronal recycling bin: Another look at huntingtin in coordinating autophagy
Thomas J. Krzystek, Shermali Gunawardena

TL;DR
This paper reviews how the huntingtin protein helps neurons manage waste through autophagy and how its dysfunction may contribute to diseases like Huntington’s.
Contribution
The paper provides a comprehensive overview of huntingtin’s role in neuronal autophagy and its implications for neurodegenerative diseases.
Findings
Huntingtin influences autophagy induction and autophagosome formation in neurons.
Huntingtin contributes to autophagosome–lysosome fusion and transport.
Dysfunction in huntingtin may disrupt autophagy, leading to neurodegenerative diseases like Huntington’s.
Abstract
Neurons, as post–mitotic and long–lived cells, rely heavily on autophagy to maintain cellular homoeostasis and ensure proper function. Huntingtin (HTT), a protein central to Huntington’s disease (HD), has emerged as a putative multifunctional regulator within the neuronal autophagy–lysosome pathway. This review explores normal HTT’s multifaceted role in neuronal autophagy, from its potential involvement in autophagy induction, its capacity to influence cargo recognition and autophagosome formation, and its contribution to autophagosome–lysosome fusion and transport. We also discuss the unique challenges that neurons face in maintaining proteostasis through autophagy, emphasising the need for specialised mechanisms like axonal transport of autophagosomes and distinct regulatory pathways. Furthermore, we highlight the spatial and temporal regulation of neuronal autophagy, particularly in…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
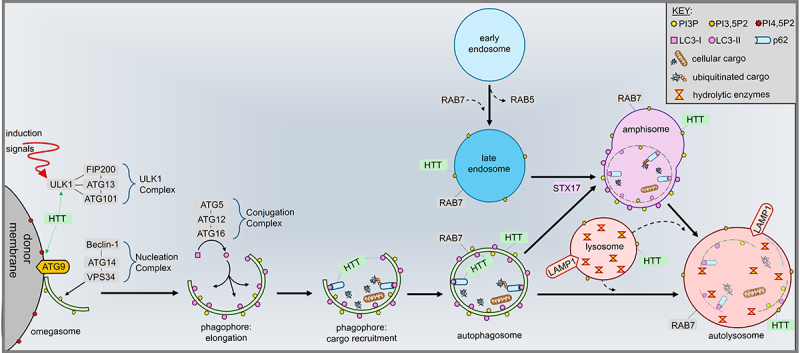 Figure 1
Figure 1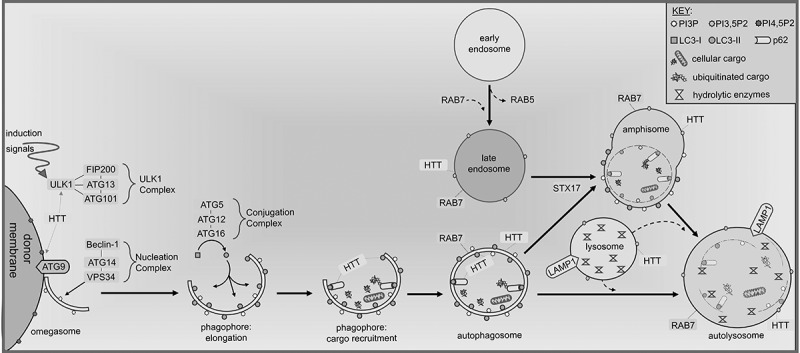 Figure 2
Figure 2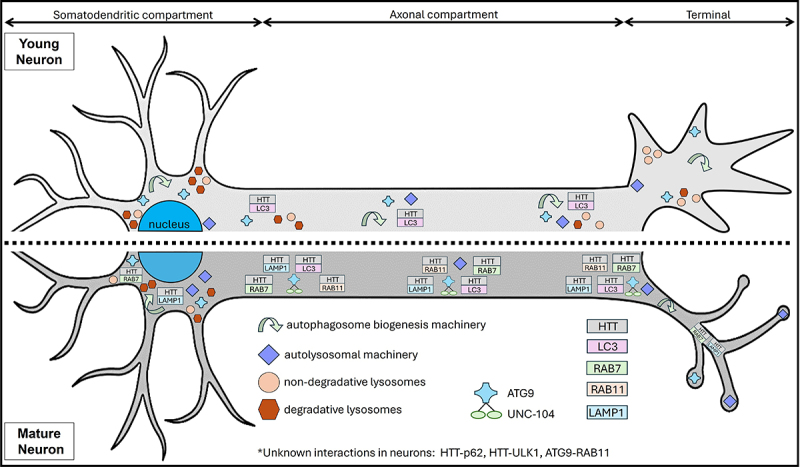 Figure 3
Figure 3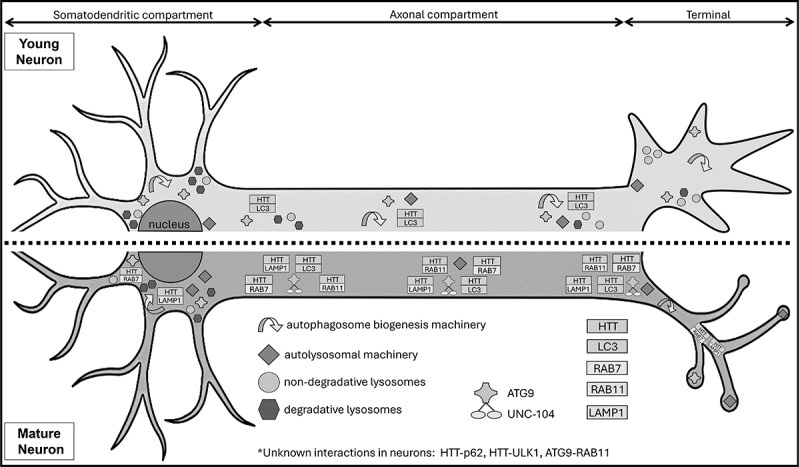 Figure 4
Figure 4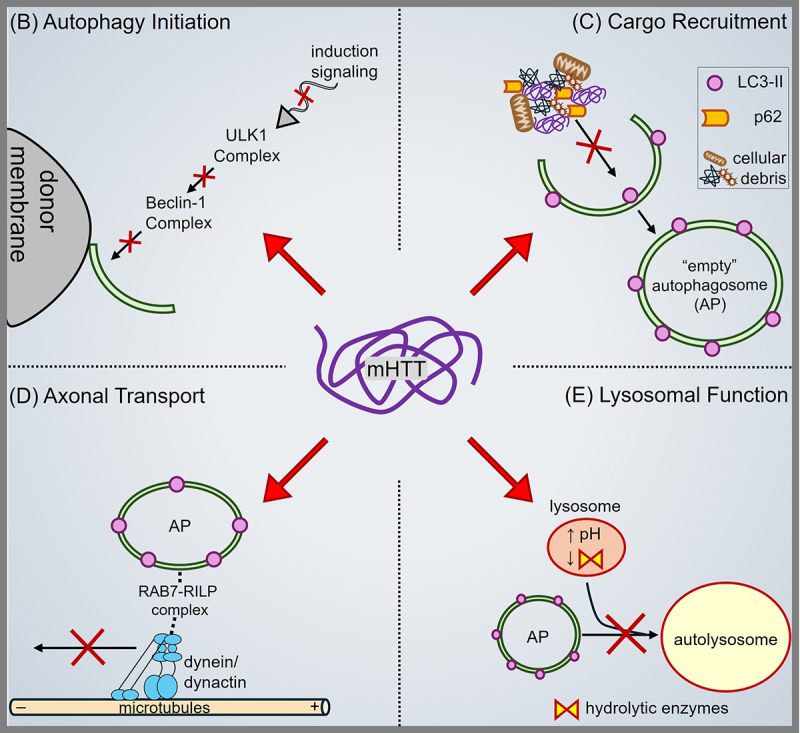 Figure 5
Figure 5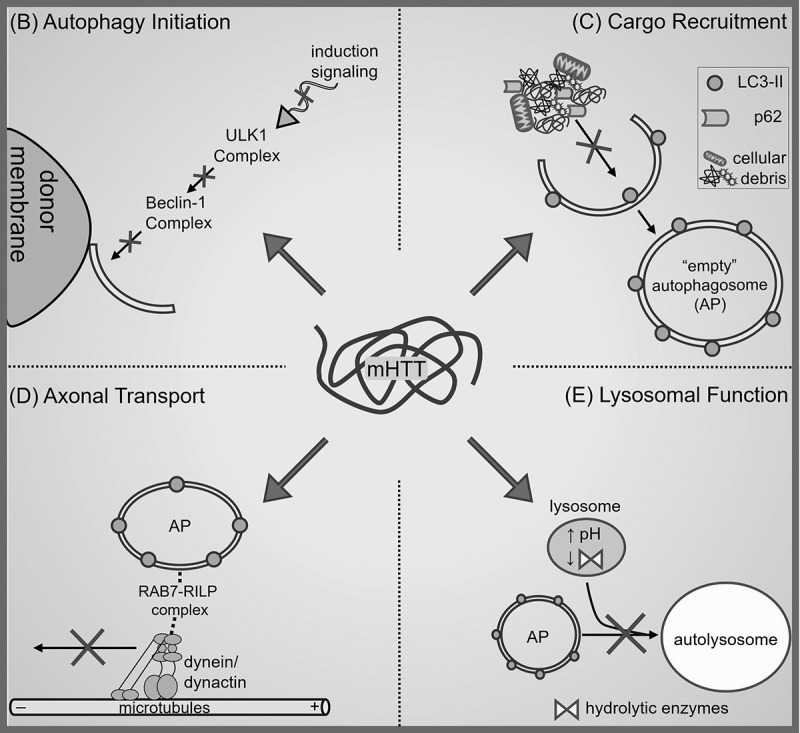 Figure 6
Figure 6Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsGenetic Neurodegenerative Diseases · Autophagy in Disease and Therapy · Mitochondrial Function and Pathology
Introduction
Neurons are post–mitotic, long–lived cells that undergo extensive metabolic demands. Unlike mitotic cells, neurons lack the ability to mitigate the accumulation of damaged proteins and organelles through cell division. Instead, they heavily depend on degradative pathways such as the ubiquitin–proteasome system [UPS, 1], the endolysosomal pathway [2], and macroautophagy/autophagy [3] to maintain cellular homoeostasis for proper neuronal health and function. Of these pathways, autophagy is a tightly regulated and highly conserved catabolic pathway across eukaryotes responsible for degrading protein aggregates, damaged organelles, and invading pathogens through a coordinated sequence of autophagy induction, phagophore elongation/assembly, cargo recruitment, and autophagosome–lysosome fusion [4]. Autophagy is essential for neuronal survival and axonal homoeostasis [5], with autophagy loss leading to neurodegeneration [6,7]. While autophagy is thought to be constitutively active in neurons under basal conditions [8], an age–related downregulation of genes essential for autophagy has been observed in the human brain [9]. Consequently, neurons experience a diminished capacity for clearing cellular debris as individuals age [10,11], likely contributing to the pathogenesis of age–related neurodegenerative disorders such as amyotrophic lateral sclerosis [ALS, 12, 13], Parkinson’s disease [PD, 14, 15], Alzheimer’s disease [AD, 16, 17], and Huntington’s disease [HD, 18, 19]. Elucidating the currently unknown regulatory mechanisms of constitutive neuronal autophagy could unlock promising new therapeutic avenues aimed at restoring neuronal functions in these deadly diseases.
HD is a devastating neurodegenerative disorder hallmarked by loss of striatal neurons [20], progressive motor dysfunction, and cognitive decline [21,22]. HD manifests from an abnormal expansion of CAG repeats (>39) in the polyglutamine (polyQ) tract of the Huntingtin (HTT) gene [23,24], with the number of CAG repeats underlying the disease onset in patients [25]. Research into HD has identified a critical role for autophagy dysfunction in disease progression. Studies in post–mortem brains of HD patients [26,27], neurons differentiated from induced pluripotent stem cells (iPSCs) from HD patients [28], transgenic mice [29,30,31], mouse clonal striatal cells [32], rat pheochromocytoma (PC12) cells [32], and fly (Drosophila) HD models [33] revealed evidence of impairments in the autophagy–lysosome pathway. Given that toxic gain–of–function [34], loss–of–function [35,36], and aberrant polyQ–mediated events likely contribute differently to HD pathogenesis, there is significant uncertainty about how autophagy defects arise in HD [18]. This ambiguity is further complicated by mounting evidence that normal or wild type HTT itself might scaffold key machinery throughout different steps of the autophagy–lysosome pathway, including autophagy induction [33], cargo loading for selective autophagy [33,37], autophagosome formation [38], autophagosome movement [39], and the potential overlap with the endolysosomal system [29,40]. In this review, we will explore the multifunctional roles of normal HTT within the autophagy–lysosome pathway.
The autophagy–lysosome pathway within neurons
Autophagy is a highly regulated cellular process that involves the degradation of damaged organelles and misfolded proteins through the formation of double–membrane vesicles called autophagosomes [4]. These autophagosomes fuse with lysosomes, where their contents are broken down and recycled, maintaining cellular proteostasis and promoting cell survival. Though key studies have laid the foundation for our understanding of the autophagy–lysosome pathway, there is still a great deal of ambiguity regarding autophagy in neurons. Here we will aim to define autophagy through the lens of a broad array of eukaryotic cells to highlight how neuronal autophagy likely faces unique challenges during autophagy induction, phagophore assembly, cargo recruitment, autophagosome fusion, transport, and maturation.
Autophagy induction
The initiation of autophagy is thought to be regulated by the mammalian target of rapamycin 1 (mTOR), which inhibits the process under nutrient–rich conditions across most mammalian cells. The core regulatory complex for autophagy induction is comprised of unc–51–like autophagy activating kinase (ULK1), focal adhesion kinase family interacting protein of 200 kDa (FIP200), AuTophaGy–related 13 (ATG13), and ATG101. Under nutrient–rich conditions, mTOR phosphorylates ULK1 at Ser757 [41] and/or Ser638 [42], thereby suppressing ULK1ʹs catalytic activity and preventing autophagy [41]. Additionally, mTOR phosphorylates ATG13 [43] which further inhibits nutrient–dependent autophagy. Conversely, rapamycin–mediated mTOR inhibition causes decreased ULK1 and ATG13 phosphorylation [42,43], triggering autophagy and the activation of other components of the ULK1 complex. ULK1 activity seems to be regulated by competing mechanisms involving either mTOR or AMP–activated kinase (AMPK) depending on nutrient availability [41]. Indeed, AMPK can phosphorylate and activate ULK1 following mTOR inhibition [41].
However, this mechanism of action for autophagy induction might require further investigations in neurons. While mouse cortical neurons exhibited nutrient deprivation–induced autophagy in an mTOR–dependent manner [44,45], mouse brain tissue did not show elevated autophagy induction in response to starvation [8,46]. However, autophagy induction upon intermittent fasting suggests mechanisms might be more nuanced in brain tissue [46]. Moreover, rapamycin does not seem to consistently induce autophagy in cultured neurons: mouse cortical neurons exhibited short–term autophagy induction with one hour of rapamycin treatment [47] but failed to demonstrate autophagy induction with 48 hours of treatment [48]. Although mTOR–dependent autophagy might play a key role in neuronal events such as synaptic pruning [49] and synaptic transmission [50], mounting evidence suggests that alternative regulatory mechanisms likely exist via kinases capable of phosphorylating ULK1, such as AMPK [51], glycogen synthase kinase 3β [GSK3β, 52], and protein kinase Cα [PKCα, 53]. Moreover, neuronal autophagy seems to be capable of induction following activation of transcription factor EB (TFEB) in an mTOR/ULK1–independent manner [54]. This body of work underscores significant gaps in our understanding of how neuronal autophagy induction is achieved and highlights a need to explore alternative pathways involved in its regulation.
Phagophore elongation/assembly
Once activated, the ULK1 complex interacts with a nucleation complex that includes Beclin–1, ATG14, and vacuolar protein sorting 34 (VPS34), a class III phosphatidylinositol 3–kinase [55,56,57], to generate phosphatidylinositol 3–phosphates (PI3Ps) essential for phagophore assembly [58]. Phagophores are a precursor to the autophagosome, and PI3Ps are also essential for recruiting proteins essential for autophagosome functions, such as WD repeat domain phosphoinositide–interacting proteins (WIPIs), Microtubule–associated protein 1A/1B–light chain 3 (LC3), and FYVE (Fab–1, YGL023, VPS27, and EEA1) domain–containing proteins [59,60]. Elongation of the phagophore is a complex process that relies on a variety of ATGs and a robust supply of lipids. Elongation involves two ubiquitin–like conjugation systems. In the first system, ATG7 (an E1–like enzyme) and ATG10 (an E2–like enzyme) conjugate ATG12 to ATG5 [61,62]. The ATG12–ATG5 conjugate, along with ATG16, acts as an E3–like enzyme to facilitate the conjugation of LC3–I (a mammalian ortholog of Atg8) to phosphatidylethanolamine (PE) to produce LC3–II [63]. This conjugation is critical for the expansion and curvature of the autophagosome membrane; however, it is relatively short lived as ATG5 and ATG12 can dissociate swiftly from the elongating phagophore following its biogenesis into an autophagosome [64]. The second conjugation system involves ATG7 and ATG3, which facilitate the processing and attachment of LC3 to PE, further aiding in membrane elongation and autophagosome maturation [65].
A vital element of autophagy is the supply of lipids for the growing autophagosome membrane. ATG9 is a protein with a transmembrane domain that plays a central role in this process by transporting lipids between organelles and the expanding autophagosome. The specialised structure at the donor membrane is referred to as the omegasome. The role of ATG9 is complemented by proteins like transmembrane protein 41B (TMEM41B), which localises to the endoplasmic reticulum (ER) and are involved in lipid transfer to the phagophore [66]. Interestingly, ATG9 has been observed to localise across a wide range of membranous structures such as ER [67], mitochondria [68], endosomes [69], golgi [70], and synaptic vesicles [71]. Unsurprisingly, a wide variety of membrane sources have been implicated in autophagy: plasma membrane [72], mitochondria [73], ER [74,75], Golgi [76], and endosomes [77]. Since ATG9–mediated lipid transfer is essential for the elongation and closure of the autophagosome, there is a need to understand how a neuron regulates the distribution of ATG9 throughout its extensive axonal network.
ER is a complex organelle canonically classified into two categories based on the absence or presence of membrane–associated ribosomes. While rough ER is ribosome–studded and involved in translation and protein–folding, smooth ER is void of ribosomes and plays more homoeostatic roles such as calcium buffering and lipid synthesis. Although rough ER has not been observed within axonal projections and is primarily localised to the somatodendritic compartment of neurons, smooth ER is thought to pervade axonal projections as narrow, continuous, and inter–connected tubules [78,79,80]. While axonal ER functions are still being explored in the context of the secretory pathway [81,82], synaptic transmission [83], and microtubule–dependent transport [84,85,86], work in mouse hippocampal neurons revealed that autophagosomes in distal neurites can form locally and colocalize with ER [64]. This supports the proposal that neurons likely utilise axonal ER as lipid donors for local autophagosome biogenesis throughout their long and complex processes; however, it remains uncertain if this is a compensatory mechanism due to the metabolic demands of neurite outgrowth following dissociation. Indeed, components of the endolysosomal system have been suggested to fuse with the plasma membrane of distal neurites to provide new lipids for extending neurite processes [87,88]. Furthermore, it remains uncertain how these protein complexes involved in phagophore elongation and assembly are moved within axons. Though loss of ATG5 in cultured mouse neurons disrupted the axonal movement of ER (labelled by the ER–localisation KDEL motif) and LC3–labelled autophagosomes [50], it is unknown how ATG5 and other members of this phagophore assembly complex are moved/distributed within axons. As we elucidate these mechanisms underlying neuronal autophagy, it is critical that distinctions between developing neurons with growth cones and matured neurons with established synapses are assessed since these states represent distinct environments with specific functional needs.
Cargo recruitment
Through the formation of double–membraned vesicles called autophagosomes, autophagy engulfs and delivers these components to lysosomes for degradation. Depending on the cargo being degraded, autophagy can be classified as either “non–selective” or “selective”. While non–selective autophagy typically involves starvation–induced bulk degradation of cytosolic components to assure adequate nutrients for cellular functions, selective autophagy involves the recruitment of specific cargo (ie: toxic protein aggregates and damaged organelles) recognised by cargo receptors [89] that are characterised by their ability to associate with autophagosomes via LC3–interacting region (LIR) motifs [90]. Though different cargo receptors have been studied in the context of different subtypes of selective autophagy (ie: mitophagy versus aggrephagy), p62/sequestosome 1 [SQSTM1, 91, 92], neighbour of BRCA1 [NBR1, 93], Tax1 binding protein 1 [TAX1BP1, 94], nuclear dot protein 52 [NDP52, 95], and optineurin [OPTN, 39] are among the more extensively studied cargo receptors for selective autophagy. In the context of neurons, work over the last decade has identified that several proteins linked to neurodegenerative diseases may associate with cargo receptors and/or LC3 [96], highlighting putative mechanisms for autophagy defects that might be neuron specific. Indeed, repeat hexanucleotide expansions in C9orf72 are the most common genetic cause of ALS [97] and C9orf72 has been observed to associate with p62 to mediate the targeting of stress granules in mouse motor neurons for p62–dependent autophagy [98]. Though it is unclear how C9orf72 recruits p62 and other autophagy machinery to help motor neurons eliminate stress granules, this work raises the question of whether this is a neuron–specific pathway to mitigate compounding stress due to a neuron’s long–lived, post–mitotic life.
Cargo receptors serve as a linker between ubiquitinated cargo, damaged organelles, and/or aggregated proteins and autophagosome–associated LC3–II, ensuring the specific incorporation of target molecules into the autophagosome. The selective degradation of ubiquitinated protein aggregates is known as aggrephagy, and p62 is the most characterised cargo receptor that is involved in recognising ubiquitin chains attached to protein aggregates [91], though NBR1 has also been shown to be involved [93]. In contrast, selective degradation of organelles, such as mitochondria, has been long thought to involve specific machinery. OPTN [39] and NDP52 [99] were shown to be critical for the selective degradation of mitochondria through a process known as mitophagy, which are recruited by the ubiquitin kinase PTEN–induced kinase 1 (PINK1) to subsequently recruit ULK1 and downstream autophagy machinery such as LC3 [99]. Though this process wasn’t thought to involve p62, there is mounting evidence that p62 may be involved in PINK1/Parkin–dependent mitophagy through a mechanism that involves NF–kB essential modulator (NEMO) which is distinct from NDP52 and OPTN [100,101]. This overlapping function of p62 highlights that these cargo receptors might not be reliable markers to distinguish between different types of autophagy and likely require tandem investigations to validate the pathways involved. Therefore, there remains a need to investigate overlapping and distinct regulatory mechanisms between the differential involvement of cargo receptors such as p62 and OPTN during selective autophagy.
Autophagosome fusion, transport, and maturation
Once an autophagosome is fully formed, it can mature by fusing endolysosomes, including late endosomes and lysosomes [102]. This fusion process is facilitated by complexes containing soluble N–ethylmaleimide–sensitive factor attachment receptors [SNAREs, 103], particularly syntaxin 17 [STX17; 104] and syntaxin 7 [STX7; 105]. In neurons, autophagosomes forming in distal neurite processes have been shown to require STX17–mediated fusion with RAB7 GTPase–containing endolysosomes, forming amphisomes, prior to retrogradely moving towards cell bodies [106]. Retrograde movement towards the soma has been shown to be critical for the maturation of amphisomes/autophagosomes in mouse hippocampal neurons [107]. However, there is conflicting evidence regarding its importance for fusion with lysosomes for degradation in mouse cortical neurons. [108,109], provided evidence that degradative lysosomes are enriched near the soma. In contrast, [110], reported that degradative endolysosomes can also be delivered to distal regions of neurites to maintain local degradation capacity. Contributing to the uncertainty of whether the soma is a primary site for lysosomal degradation in neurons is the use of markers for these organelle structures that might not be as discrete as originally thought. LC3, the primary marker used to monitor the dynamics of autophagosomes [111], is present across multiple phases of the autophagy pathway beginning with phagophore assembly and ending with lysosomal fusion. Likewise, lysosome–associated membrane protein 1 (LAMP1), the predominant marker used to monitor lysosome dynamics, was shown in mouse cortical neurons to label a significant proportion of organelles that do not contain the hydrolytic enzymes and proteases necessary for degradation [108]. Therefore, while we need to be cautious about concluding the identity of organelles using these markers, there is a need to identify additional components of these autophagic and lysosomal structures to distinguish the specific compartments being investigated.
Late endosomes are predominantly characterised by the presence of RAB7–GTPase responsible for the coordination of late endosomal functions [112,113]. Although fusion of RAB7–containing endolysosomes with LC3–containing autophagosomes have been shown to be critical for retrograde movement and maturation, RAB7 can coordinate both the anterograde and retrograde movement of late endosomes/endolysosomes. During retrograde axonal movement, GTP–bound RAB7 can interact directly with RAB7–interacting lysosomal protein (RILP) at its effector–interacting switch region [114,115]. The RILP–RAB7 association has been shown to prevent RAB7 from hydrolysing GTP, thereby keeping it in the active state that is vesicle bound [115]. RILP localises to RAB7 by binding the VPS41 subunit of the homotypic fusion and protein sorting (HOPS) complex [116]. RILP was shown to aid the recruitment of dynein/dynactin to RAB7–containing membranes through direct interaction with the p150glued C–terminus [117]. Moreover, oxysterol–binding protein–related protein 1 L (ORP1L) simultaneously binds RAB7 and RILP to stabilise their interactions with dynein/dynactin [117,118,119]. Alternatively, RAB7 mediates anterograde movement through the recruitment of the FYVE and coiled–coil domain–containing protein 1 [FYCO1/RUFY5; 120]. FYCO1 serves as a coincidence detector that can interact with both RAB7, PI3Ps, and LC3 [86,120], highlighting a potential mechanism in which RAB7–containing endolysosomes recruit LC3–containing autophagosomes. Additionally, FYCO1 has been shown to aid the recruitment of kinesin–1 to RAB7–containing membranes through direct interaction with kinesin–1 [120]. Although FYCO1–dependent anterogradely moving RAB7–containing endolysosomes were observed in projections of PC12 cells [86], the biological role of an anterogradely moving RAB7–containing vesicle within a neuron remains unclear. Perhaps these are endolysosomes contributing to the local degradation capacity in distal neurites [110], but further work will be needed to test these predictions in the context of a neuron.
Normal, wild type huntingtin (HTT) as a multifunctional scaffold during autophagy
HTT is a very large 350 kDa protein that is ubiquitously expressed but enriched in the brain [121,122]. Though researchers identified HTT over thirty years ago to be the genetic cause of HD [123], the normal function of HTT within neurons remain elusive despite being essential for development with loss of HTT causing early embryonic lethality [124,125]. One proposed function of HTT is as a multifunctional scaffolding protein at membranes based on key components of its structure as well as its numerous interacting partners. Indeed, HTT has several major HEAT (Huntingtin, Elongator factor3, PR65/A regulatory subunit of PP2A, and Tor1) repeat domains, giving a solenoid–like shape [126], believed to enhance protein–protein interactions and scaffolding functions [127,128]. The sweeping presence of these HEAT repeat domains across the length of HTT supports scaffolding functions near HTT’s N– and C–terminal domains [129]. Moreover, the N–terminus of HTT exhibits a proline–rich region that enhances structural flexibility and aids protein–protein interactions [130]. Further evidence for a putative scaffolding role is the fact that HTT interacts with more than 350 partners isolated through yeast–two hybrid [Y2H; 131, 132] and mass spectrometry (MS) proteomics [133,134,135]. However, the biological relevance of this myriad of interactions remains unknown likely due to HTT’s complex cellular localisation pattern across membrane–bound organelles.
Biochemical analysis showed HTT to be at membranes [136], and HTT can associate with membrane phospholipids [137,138,139], likely by an amphipathic alpha helical membrane–binding domain in the first 17–18 amino acids of HTT’s N–terminus [139,140,141,142]. Membrane–bound HTT can function in a variety of cellular processes including neural adhesion [143], endocytosis [144], vesicle fusion [146], and transport [147,148]. Indeed, HTT localises to a variety of membranous subcellular compartments such as synaptic [136] and RAB–containing [40,149,150,151] vesicles, plasma membranes [139], ER, Golgi, endosomes [140,152], and autophagosomes [39]. Moreover, deletion of the N–terminal alpha helical structure in HTT resulted in decreased HTT localisation across ER, golgi, and vesicle membranes, increased nuclear accumulation, and induced cytotoxicity [140,153], highlighting the importance of these first 17–18 amino acids for HTT’s subcellular localisation across membranes.
HTT may preferentially associate with membranes depending on the local populations of phosphoinositide (PI) phospholipids, with a strong bias for phosphatidylinositol 3,5–bisphosphate (PI3,5P2) and phosphatidylinositol 4,5–bisphosphate (PI4,5P2) observed in vitro [139]. Endolysosomes are heavily decorated with PI3,5P2 [154,155], which are produced from PI3Ps by PIKfyve in mammals [156]. Loss of PIKfyve, and subsequently PI3,5P2 was observed to be embryonically lethal in knockout mice [157]. Although PIKfyve and PI3,5P2 are critical for the distribution and motility of endolysosomes in human cells and mouse fibroblasts [158], their regulation, localisation, and downstream effect on the autophagy–lysosome pathway in neurons remain obscure. Since reliable live–cell reporters for PI3,5P2 have only recently been discovered [159], we are now better equipped with tools to help clarify how PI3,5P2 helps coordinate neuronal autophagy and how PI3,5P2–containing organelles are regulated within the complex architecture of a neuron. Although PI4,5P2 are heavily produced and localised at plasma membranes [160], they have also been identified at other organelles such as autophagosomes [161], perhaps acquired through fusion with endolysosomes following endocytosis from the plasma membrane [162,163]. While PI4,5P2 has been proposed to play a role in upstream autophagy induction – before PI3Ps – by regulating the recruitment of ATG14 [164], PI4,5P2 has been well studied in the context of autophagosome fusion with lysosomes [165]. Indeed, disrupted generation of phosphatidylinositol 4–phosphate (PI4P) resulted in defective lysosome–autophagosome fusion via RAB7 inactivation in human HEK293 cells [166]. Although HTT has been shown to form a complex with RAB7 on endolysosomes in mature fly axons Figure 1; [40, 150], the upstream events that dictate HTT’s localisation across these membranous compartments remain unclear. With recent advances in our understanding of PI3,5P2 and PI4,5P2 in the autophagy–lysosome pathway, it is also possible to predict that these unique lipids can help recruit HTT to these organelles (Figure 1). Figure 1.Schematic representation of HTT’s multifunctionality in the autophagy–lysosome pathway. HTT interacts with ULK1, aiding its recruitment to donor membranes containing PI4,5P2 and ATG9, leading to phagophore nucleation, and LC3–I conjugation to LC3–II. HTT can interact with p62 for cargo recruitment of cellular debris, damaged organelles, and ubiquitinated proteins. Subsequently, HTT associates with autophagosomes and late endosomes, likely acting in concert with RAB7 for microtubule–dependent transport to lysosomes following STX17–mediated fusion. Finally, HTT is implicated in lysosome fusion with both autophagosomes and amphisomes for the degradation of cellular cargo.
HTT’s localisation and membrane trafficking can also be regulated by post–translational modifications, including palmitoylation at Cys214 [167]. Interactions with the palmitoyl–acyl transferases (PATs) HIP14 (ZDHHC17) and HIP14L (ZDHHC13) facilitate HTT palmitoylation [167]. Furthermore, HTT may also play a role in regulating HIP14/HIP14L–mediated palmitoylation of other proteins since reduced HIP14 PAT activity was observed in mice exhibiting an antisense oligonucleotide–mediated HTT knockdown [169]. Further evidence in Y2H supports this, revealing a strong overlap between HTT and HIP14 interactors [170]. While palmitoylation helps regulate the localisation of both normal and pathogenic HTT [167], as well as the localisation of pre–synaptic vesicle proteins such as SNAP25 and GluR1, [170, 169, 171, 172], it remains to be tested whether HTT–mediated HIP14 PAT activity plays a role in the autophagy–lysosome pathway. Be that as it may, HIP14L (ZDHHC13) was observed in HeLa cells to palmitoylate ULK1 and siRNA–mediated reduction of ZDHHC13 showed disrupted autophagic flux and decreased abundance of LC3–containing cargo [173]. Therefore, it is possible that HTT palmitoylation and/or HTT–mediated HIP14/HIP14L PAT activity plays a key role in regulating the autophagy–lysosome pathway. Although overexpression of HIP14 in mice expressing pathogenic HTT has shown therapeutic promise [167], it remains to be tested whether this rescue involves alleviation of mutant HTT (mHTT)–mediated autophagy defects.
Over the last decade, conflicting evidence hints at a multifaceted role for HTT in the autophagy pathway. Work in fly and human (HEK293) cells revealed that HTT interacts with ULK1, implicating it in the upstream induction of autophagy Figure 1; [174, 33]. Furthermore, HTT may be involved in cargo recruitment through observed interactions with p62 [33,174] and LC3 [37] to facilitate the coordination of materials to the elongating phagophore (Figure 1). However, the subcellular localisation of HTT–ULK1 and HTT–p62 associations were not examined in neurons. In fly neurons with functional synapses, HTT was not observed to co–localise with ATG5 in axons [40]. This implicates that HTT’s role in axons might be distinct from its role with ULK1 in autophagy induction and with p62 in selective autophagy – perhaps at axon terminals and at the soma. Additionally, HTT interactions with HAP1 and the molecular motor proteins kinesin–1 and dynein/dynactin [130,148] supports a role in trafficking of autophagosomes and endolysosomes Figure 1; [39], likely to ensure their proper distribution within growing neurons. However, whether this is the case in a mature neuron is unknown. Furthermore, expression of excess HTT in mice revealed increased endolysosomal activity [29], highlighting HTT as supporting lysosomal clearance pathways. Moreover, structural properties of the HTT protein shares considerable similarities with three essential autophagy–related proteins: Atg23, Vac8, and Atg11 [175]. Additionally, a caspase–3–cleaved fragment of HTT was found to be similar to an autophagosome–targeting sequence domain of ATG14L shown to regulate autophagy by sensing and promoting membrane curvature [18]. Collectively, these reports implicate HTT as a key player in neuronal autophagy, orchestrating various stages of this essential cellular process (Figure 1).
Challenges for autophagy in neurons and HTT’s physiological role
Being post–mitotic, neurons face unique challenges in maintaining cellular homoeostasis through autophagy. The task of degrading and recycling damaged proteins and organelles is further complicated by the extensive length of neuronal axons, which can extend up to 1 mm in humans. Further, the maintenance of cellular homoeostasis during neuronal growth where components need to be degraded quickly in contrast to established neurons can add to the complexity. Unlike mitotic cells, neurons likely adapted autophagic machinery and regulatory pathways to overcome these challenges. Overall, the neuron must conquer four major hurdles in the context of autophagy. These are (1) axonal transport of autophagosomes, (2) the distinct regulatory mechanisms of neuronal autophagy induction, (3) the spatial production of autophagosomes within a neuron, and (4) the temporal regulation of autophagy in an ageing neuron.
Axonal transport of autophagosomes
Although autophagosomes are proposed to form distally in axons [72,176], degradative lysosomes capable of hydrolysing proteins and damaged organelles are reported to be enriched near the soma [108,109]. Even though evidence in mouse cortical neurons suggests that degradative lysosomes anterogradely traffic to distal axons to maintain local degradation capacity [110], disrupting the retrograde movement of autophagosomes and endolysosomes resulted in the aberrant accumulation of these cargo in axons [106]. This suggests that the local degradative capacity may not be sufficient for clearance of autophagy cargos and that retrograde movement to the soma is necessary for other events in the pathway such as recycling via Golgi [178]. It is well characterised that autophagosomes acquire machinery for retrograde movement via fusion with endolysosomes by STX17 [106]. HTT has been implicated in supporting the long–distance trafficking of various cargoes, including synaptic vesicles [148], endosomes [150,151], and autophagosomes [39]. Through associations with Huntingtin–associated protein 1 (HAP1) and the dynein/dynactin complex [179,180], HTT helps coordinate the movement of autophagosomes generated at the distal ends of axons towards the cell body for fusion with lysosomes [39]. HTT is also involved in forming a complex with RAB7 on retrogradely moving endolysosomes that contained STX17 [40], Figure 1. Despite this knowledge, the precise mechanisms coordinating the recruitment of autophagosome–endolysosome fusion machinery in distal axons remain unclear. Evidence suggests that autophagosomes and amphisomes may use sequential adaptors, such as c–Jun N–terminal kinase–interacting protein 1 (JIP1), c–Jun N–terminal kinase–interacting protein 3 (JIP3), and HAP1, during retrograde trafficking [181], but the mechanisms for their differential recruitment are unknown. Interestingly, work in PC12 cells indicated that endolysosomes repeatedly contact axonal ER during transit within neurite projections to coordinate exchanges of motor proteins and adaptor proteins that help drive motility [86]. Furthermore, lysosome contacts with ER near the soma appear to be required for lysosomal transport within axons [182], further supporting a regulatory link between ER and the autophagy–lysosome pathway. However, while the relevance of this mechanism to autophagosomes needs to be tested, HTT is also capable of associating with ER [140], making it a promising candidate to explore in mediating the exchange of motor proteins and adaptors between endolysosomes/autophagosomes and the axonal ER. Taken together, HTT appears to play a critical role in coordinating the retrograde movement of autophagosomes and endolysosomes, but further explorations are needed to test if HTT also coordinates the scaffolding of the SNARE fusion machinery responsible for autophagosome fusion with endolysosomes. Moreover, precise markers specific to different endolysosome and autophagosome cargoes which are currently lacking (for e.g., amphisomes versus autophagosomes, catalytically competent endolysosomes,) would aid in identifying the exact steps in the autophagy–lysosome pathway.
Regulatory mechanisms of neuronal autophagy induction
Autophagy in neurons exhibit unique regulatory characteristics compared to non–neuronal cells, notably showing less susceptibility to mTOR regulation [47]. Neurons rely on highly active constitutive autophagy under basal conditions [8]. However, the upstream pathways that activate ULK1–mediated autophagy in neurons, or potential alternative pathways, remain to be fully understood. Being post–mitotic, neurons likely adapted multiple pathways that can trigger clearance of damaged proteins and organelles in response to intrinsic and extrinsic stressors. Since we do not have a clear picture of the diverse autophagy induction pathways within neurons, it is difficult to understand how these might go awry and contribute to neurodegenerative diseases, including HD. While HTT interacts with ULK1 independent of the mTOR–ULK1 complex in fly and human cells [33,174], whether similar associations also occur in neurons is unclear. Therefore, it is unknown if HTT has a role during neuronal autophagy induction. Loss of HTT reduced autophagy activity in fly neurons [37] as well as mouse embryonic fibroblasts (MEFs), N2a neuroblastoma cells, and striatal cells [33]. However, mice expressing excess normal, non–diseased, wildtype HTT exhibited enhanced autophagy activity [31]. In contrast, other work failed to find HTT co–localising with ATG5 in axons of fly neurons [40]. While it is unclear if the HTT–ULK1 complex functions in ATG5–independent pathways, it is possible that HTT could play a specific role initially prior to autophagosome assembly. Alternatively, certain small molecules can induce autophagy in neurons, providing a clue to the involvement of other kinases in upstream signalling pathways. For example, 10–[4′–(N–diethylamino)butyl]–2–chlorophenoxazin (10–NCP), an AKT inhibitor, has been shown to enhance autophagy in mouse striatal, cortical, and hippocampal neurons [48]. 10–NCP was further shown to enhance the retrograde axonal transport of late endosomes and autophagosomes in cortical neurons expressing mutant human amyloid–beta precursor protein [APP; 183], supporting a link between retrograde axonal transport and autophagy activity. While it is unknown if HTT is involved in this same pathway, work on 10–NCP opens an exciting avenue to explore other kinase pathways that may be involved during autophagy induction. Further, it would also be interesting to explore how the HTT–ULK1 complex changes in the presence or absence of autophagy inducing agents such as 10–NCP to determine if this complex is a requirement for neuronal autophagy activity.
Spatial production of autophagosomes within a neuron
Due to their complex architecture, neurons are faced with a unique issue since cargo that needs to be degraded can be in distal processes, far from the soma where a lysosomal machinery is predominantly concentrated. This necessitates a highly coordinated and localised generation of autophagosomes within the distal regions of the neuron to ensure efficient degradation of cellular components. Localised autophagosome generation allows for the immediate encapsulation and degradation of cargo, preventing the accumulation of potentially toxic materials like protein aggregates and damaged organelles. ATG9–mediated phagophore assembly has been implicated across a wide range of membranous compartments [184] and ATG9–containing vesicles have been shown to contribute to autophagosome formation [185]. Though ATG9–containing vesicles were identified from subcellular fractionation of rat brain tissue, it remains unclear how ATG9 is trafficked, distributed, and activated within distal axonal processes. ATG 9 was shown to be enriched at nerve terminals, therefore ATG9 must be transported from the soma to the synapse Figure 2, [186]. Vesicular ATG9 was also observed to move anterogradely in C. elegans with RAB3 by the aid of KIF1A/UNC–104 which transports synaptic vesicles [71], and HTT has been observed to facilitate the axonal transport of RAB3–containing vesicles in fly axons [150]; however, it is unknown if this putative HTT–RAB3 vesicle overlaps with ATG9–RAB3 vesicles. Moreover, these ATG9 vesicles may also undergo exo/endocytosis at the pre synapse in response to stimulations, similar to what is observed for Rab11 and HTT containing vesicles [187]. Figure 2.Spatial and temporal distributions of selected machinery in the autophagy–lysosome pathway. In a young neuron with an active growth cone, degradative lysosomes are primarily localised within the somatodendritic compartment, while non–degradative lysosomes are more prevalent in axons and terminals. Autophagosome biogenesis machinery, including ATG9, and autolysosomal components are distributed across the neuronal compartments, though the precise mechanisms of their trafficking remain unclear. In axons, HTT and LC3 co–transport, suggesting a coordinated role in autophagy. In a mature neuron with a fully developed synaptic terminal, both degradative and non–degradative lysosomes are present within the somatodendritic compartment, although their precise distribution in axons and terminals remains less defined. Autophagosome biogenesis machinery is present in the somatodendritic compartment and axon terminals, but their specific distribution and mechanisms of transport within axons are still unclear. In contrast, autolysosomal machinery is distributed throughout the depicted neuronal compartments, yet the mechanisms governing their transport within axons are not fully understood. In mature neurons, ATG9 vesicles are transported within axons by UNC–104, while HTT is observed to move within axons in association with RAB7, LAMP1, LC3, or RAB11.
Interestingly, the HTT N–terminus has been proposed to maintain similarity with ATG23 [175], an upstream autophagy protein critical for ATG9 trafficking in yeast [188]. Isolation of ATG9–containing vesicles from rat brain tissue suggests that HTT may be present with these vesicles [186] and a BiolD proteomics analysis of ATG9 interactors in HEK293 cells revealed HTT, HIP1, and 40–kDa huntingtin–associated protein [HAP40; 189]. However, it remains unknown if HTT directly interacts with ATG9 and/or if it regulates the distribution of ATG9–containing vesicles within neurons. Although ATG9 localises with RAB11–containing vesicles in HEK293 cells, and HTT has been shown to mediate the axonal transport of RAB11–containing vesicles in fly axons [149], it is uncertain if the RAB11–ATG9 and RAB11–HTT vesicle complexes are overlapping in the context of autophagy.
Further, the recruitment of ATG9 to ULK1 [190] remains poorly studied in neurons, leaving speculation that perhaps the HTT–ULK1 complex [33,174] is playing a vital role coordinating this recruitment. Indeed, the recruitment of ATG9 to ULK1 has been suggested to be dependent on adaptor protein complex 2 (AP–2) via interactions with TBC1 Domain Family Member 5 [TBC1D5; 191], a major component of clathrin–mediated endocytosis [CME, 192]. In neurons, CME can involve PI4,5P2 [193], huntingtin–interacting protein 1 [HIP1, 194] and dynamin 1 [195]. Independent studies have shown that HTT interacts with all these components [139,196,197] and AP–2 interacts with HIP1 [194,198]. However, it is unknown if HTT plays a role during CME/endocytosis and whether such a role helps coordinate ULK1 and ATG9 in the autophagy–lysosome pathway in neurons and particularly in the distal regions of the axons.
Temporal regulation of autophagy in an ageing neuron
In humans, neurons project long axons that innervate target cells up to 1 metre away from their cell body. Young neurons, characterised by active growth and high plasticity, exhibit robust axon outgrowth driven by dynamic growth cones at their tips [199]. These growth cones, rich in actin and microtubules, navigate the extracellular environment by responding to various guidance cues, facilitating rapid synaptogenesis. In contrast, mature neurons focus on maintaining established synaptic connections, displaying reduced plasticity and a more stabilised cytoskeleton. This transition involves a shift in molecular composition, with decreased expression of growth–associated proteins and increased stabilisation of the cytoskeleton through neurofilaments and synaptic proteins [200]. The use of adult neurons in culture from mice or rats is challenging due to their inherently poor axon regenerative capacity, influenced by both intrinsic downregulation of growth–promoting genes [201,202]. Consequently, researchers often utilise embryonic neurons from mice or rats for their superior axon outgrowth capabilities [203], despite these cells not fully representing a mature neuronal state. Furthermore, cultured neuronal systems predominantly study actively growing axons with growth cones, focusing on mechanisms of axon guidance and extension, whereas the dynamics of terminally synapsed axons, which involve the maintenance and function of established synaptic connections, are less frequently explored. Additionally, the use of human iPSC–derived neurons poses challenges as they lose the age/maturation characteristics of the individual donor, due to the reprogramming process that reverts fibroblasts to a pluripotent state [204,205]. While ongoing research aims to induce ageing [206] or directly differentiate human fibroblasts into neurons [207], bypassing the need to generate stem cells first, the metrics for accurately modelling aged or mature human neurons in culture remain non–standardised. This highlights the ongoing difficulty in creating in vitro models that accurately reflect the physiological and pathological states of mature human neurons. In contrast, model systems such as Drosophila and C. elegans can help overcome this gap in the field as in vivo models of neurons with long axons with terminal synapses.
Such challenge surfaces when studying proteins such as HTT. Loss of HTT in mice leads to embryonic lethality by embryonic day 7.5 [124,125]. However, conflicting evidence leaves the importance of HTT in adult mice to be controversial. While 35,showed that loss of HTT caused neuronal/axon degeneration and early mortality in 8–month–old mice, 208,found that loss of HTT in adult mice (4– & 8–month–old) exhibited normal lifespans. Reduced HTT in adult mice was shown to cause abnormalities such as increased striatal neuropathy [209]. Similar to HTT, the distinctions between mechanisms driving autophagy in young neurons versus mature neurons has scarcely been examined despite being essential for both neuronal development and neuronal survival/homoeostasis [210]. Interestingly, loss of autophagy was shown to affect lifespan in C. elegans, Drosophila, and mice – with key genes in the autophagy–lysosome pathway being observed to undergo an age–dependent decline in expression. Investigating how ageing and maturation of neurons influence autophagy and the proteins involved, including HTT, is crucial for understanding the distinctive roles these process is affected at different life stages. Elucidating the differences in autophagic mechanisms between young and mature neurons will also allow researchers to gain insights into how to enhance autophagy therapeutically in the context of neurodegeneration, in established neuronal populations.
Autophagy in HD
HD is a devastating, dominantly inherited neurodegenerative disorder that manifests from an N–terminal CAG repeat expansion in the HTT gene. Elucidating mechanisms underlying mHTT–mediated autophagy defects is challenging due to evidence that HD progression derives from gain–of–function [34], loss–of–function [35,36], and/or aberrant polyQ–mediated events. While growing evidence supports a role for normal, wildtype HTT in the autophagy–lysosome pathway [33,39], HD transgenic mice showed autophagosome–like vacuole accumulations prior to detectable disease onset [211], suggesting autophagy defects might underscore HD pathogenesis (Figure 3). Below, we will discuss how pathogenic mHTT can lead to (1) defective autophagy initiation, (2) impaired cargo loading of autophagosomes, (3) perturbed axonal transport of autophagy–lysosome compartments, and (4) disrupted lysosomal functions. This multi–faceted dysregulation of autophagy in HD highlights the need for therapeutic strategies that can restore autophagy activity and enhance clearance of toxic mHTT aggregates, potentially mitigating neuronal loss and disease progression. Figure 3.Pathogenic HTT disrupts the Autophagy–Lysosome Pathway in HD. Pathogenic mHTT has been evidenced to cause multi–faceted disruption of the autophagy–lysosome pathway, leading to (A) defective induction signalling for autophagy initiation, (B) disrupted cargo recruitment during autophagosome formation leading to “empty” autophagosomes and accumulations of cellular debris in the cytosol, (C) perturbed axonal transport of autophagy–related compartments such as autophagosomes and lysosomes, and (D) impaired lysosomal function underscored by deficient acidification, reduced abundance of lysosomal hydrolytic enzymes, and improper autophagosome–lysosome fusion.
Defective autophagy initiation
Post–Mortem analyses of brains from HD patients revealed accumulations of endolysosomal vesicles [26,27]. However, observations in HD transgenic mice show reduced autophagy–mediated degradation with mHTT expression as indicated by accumulated LC3–II and p62 [30]. Dysregulation of autophagy initiation is one putative mechanism that underscores this decoupling of autophagy initiation from a cell’s degradative capacity (Figure 3A). Work in HEK293 cells suggested that normal, non–pathogenic HTT facilitates mTOR activity by mediating an interaction between mTOR and Rheb, a small G protein, which becomes exacerbated with mHTT expression leading to increased mTOR1 activity [212]. Though we previously discussed the controversial effect of rapamycin on neuronal autophagy, an alternative small molecule inhibitor of mTOR1, CCI–779, was identified to reduce mTOR activity and decrease mHTT aggregates in transgenic HD mice [213]. Parallel to mTOR hyperactivity, AMPK activation has been suggested to be neuroprotective in transgenic HD mice, with potential therapeutic avenues involving the AMPK–activating metformin [214]. However, the role of AMPK in HD pathogenesis is likely complex since later work in HD transgenic mice found that chronic AMPK hyperactivation exacerbated HD pathogenesis with increased mHTT aggregates and increased cell death [215]. Interfering with AMPK pathways via CGS21680, an A(2A) adenosine receptor (A(2A)–R)–selective agonist [216], alleviated the detrimental effects of AMPK hyperactivation in HD transgenic mice [215]. Downstream of autophagy signalling, work in HD transgenic mice suggests differential dysregulation of ULK1 kinase activity with decreased phosphorylation of Beclin–1 and ATG14 [217] but increased phosphorylation of p62 [218]. Moreover, mHTT aggregates sequester Beclin–1 [33]. Since ATG14 phosphorylation [217] and Beclin–1 disinhibition [219] may enhance mHTT clearance, therapeutic efforts targeting activation of ATG14, Beclin–1, and other ULK1 targeting proteins may curtail the complexity surrounding upstream components such as mTOR and AMPK.
Impaired cargo loading of autophagosomes
While studies in human and rodent HD samples reveal an increased abundance of autophagosomes, these are often functionally impaired, “empty” autophagosomes that lack cargo to be degraded [Figure 3B; 30, 220]. Though biochemical analysis identified that mHTT aberrantly interacts with and sequesters p62 [30], the precise mechanism(s) driving this “fail–to–detect” model remain unclear. In contrast, a “fail–to–engulf” model has recently been proposed in which p62 properly detects cargo, but autophagosomes fail to properly engulf the p62–detected cargo through mechanisms involved during phagophore assembly [221]. Since it is possible that defective autophagosome engulfment and cargo detection can contribute to the formation of “empty” autophagosomes in HD [220], this complicates therapeutic interventions aimed solely at upregulating autophagy and suggests a need for avenues that enhance cargo recruitment/engulfment. A recently developed technology called AUTOTAC (AUTOphagy–Targeting Chimera) uses small peptide agonists that bind and activate p62 to facilitate the selective degradation of cargo through p62–dependent autophagy [222]. Ji et al used AUTOTAC to selectively target mHTT expressed in HeLa cells where they observed increased recruitment of mHTT to LC3–positive compartments as well as increased degradation of mHTT. While AUTOTACs represent a promising platform for targeted protein degradation in HD, critical questions remain regarding their pharmacological properties, catalytic potential, and selectivity. Future studies are needed to optimise their design, address off–target effects, and fully elucidate their mechanisms of action, including the effect of p62 sequestration on target degradation.
Perturbed axonal transport of autophagy–lysosome compartments
Pathogenic mHTT with aberrant polyQ–expansion exacerbates autophagy dysfunction by disrupting the axonal transport of autophagosomes Figure 3C; [39] and endolysosomes [223]. Previously, we discussed that autophagosomes forming in distal neurite processes require STX17–mediated fusion with RAB7–containing endolysosomes prior to retrograde axonal transport [106]. Recent work in Drosophila identified that HTT and STX17 co–localise on axonal endolysosomes containing RAB7 [40]. One possible mechanism driving impaired axonal transport of autophagosomes is that pathogenic mHTT disrupts STX17–mediated fusion of autophagy–lysosome compartments. Furthermore, mHTT has been shown to aberrantly interact with HAP1 [224], and HTT–HAP1 associations are critical for the retrograde axonal transport of autophagosomes [180,181]. Therefore, it is also possible that mHTT leads to discoordination and improper recruitment of dynein motor proteins and regulators involved in retrograde axonal transport. Similarly, pathogenic mHTT caused a weakened interaction between RILP and dynactin p150Glued, a co–factor protein associated with dynein [225]. Moreover, mHTT also impaired a complex containing OPTN and RAB8 responsible for lysosomal trafficking that resulted in decreased lysosomal activity and a lower lysosomal content of proteases like cathepsin D [226]. Taken together, targeting complexes containing STX17, RILP, or OPTN/RAB8 would be promising candidates to directly alleviate mHTT–mediated transport dysfunction in the autophagy–lysosome pathway.
Disrupted lysosomal functions
Studies in iPSC–derived neurons from HD patients revealed impaired autophagosome–lysosome fusion and reduced clearance of mHTT aggregates Figure 3D; [28]. Additionally, mHTT can impair lysosomal function by altering lysosomal pH and inhibiting lysosomal enzymes Figure 3D; [29]. Trehalose is a natural disaccharide that stimulates lysosomal biogenesis by causing lysosomal enlargement and low–grade lysosomal stress that leads to activation of TFEB [227]. Trehalose was successful in HD pathogenesis in transgenic HD mice [228]. Since trehalose also disrupts lysosomal acidification [227], there is concern that chronic application of trehalose may lead to unwanted secondary effects in cells less vulnerable to pathogenic mHTT in HD. Complimentary to this approach would be the use of acidifying nanoparticles to restore lysosomal acidification in HD. Acidifying nanoparticles have been shown to traffic to lysosomes and promote acidification in ARPE–19 cells [229], pancreatic β–Cells [230], and dopaminergic neurons from a mouse model of Parkinson’s disease [231]. Perhaps a combination of trehalose and acidifying nanoparticles could provide a dual–targeted approach to improve lysosomal function and mitigate the effects of mHTT aggregation in HD.
Extracellular vesicles have emerged as a compensatory mechanism, facilitating the secretion and removal of mHTT aggregates when the autophagy–lysosome pathway is dysfunctional [232]. The autophagy–lysosome pathway and extracellular vesicles (EVs), including exosomes, exhibit a dynamic interplay critical for maintaining cellular homoeostasis [233]. Notably, mHTT has been detected in EVs isolated from transgenic and knock–in porcine models of HD, as well as in the plasma of HD patients [234]. By encapsulating and transporting mHTT aggregates to extracellular spaces or neighbouring cells, EVs can reduce the intracellular burden of toxic mHTT aggregates. However, this mechanism also carries the risk of contributing to non–cell–autonomous pathology, as the prion–like propagation of mHTT aggregates may exacerbate disease progression through cell–to–cell transmission [235]. Understanding the dual role of EVs in mitigating intracellular toxicity and potentially driving intercellular pathology underscores the therapeutic potential of targeting the autophagy–EV axis in HD. Modulating these pathways may offer innovative strategies to enhance aggregate clearance while limiting their pathological spread, presenting a promising avenue for therapeutic development.
Concluding remarks
In summary, this review illuminates the intricate relationship between HTT and neuronal autophagy. The emerging consensus is that HTT plays a multifaceted role in this process, influencing various stages from induction to cargo recruitment and transport. HTT’s scaffolding properties enable interactions with key autophagy machinery, including ULK1, p62, HAP1, and molecular motors like kinesin–1 and dynein/dynactin. These interactions likely facilitate the coordination of autophagosome biogenesis, cargo loading, and trafficking (Figure 1), thus influencing neuronal homoeostasis and function. However, the precise mechanisms governing these interactions, and the specific subcellular locations where they occur, are still uncertain and need to be investigated.
Neurons, with their unique challenges in maintaining cellular homoeostasis due to their post–mitotic nature and extensive axonal networks, rely on a finely tuned autophagic system. The distinct regulatory mechanisms of neuronal autophagy induction, spatial production of autophagosomes, and axonal transport mechanisms highlight the specialised nature of this process (Figure 2). Moreover, the temporal regulation of autophagy in ageing neurons underscores the need to differentiate between mechanisms in young, growing neurons versus mature neurons with established synapses (Figure 2). Future studies using diverse model systems, including aged human iPSC–derived neurons and in vivo models such as Drosophila, C. elegans, and mice will be crucial to unravelling these complexities.
The potential therapeutic implications of modulating neuronal autophagy in neurodegenerative diseases such as HD cannot be overstated (Figure 3). A deeper understanding of the precise mechanisms by which HTT influences neuronal autophagy, in conjunction with the development of specific markers for different autophagic compartments, will likely pave the way for targeted interventions aimed at restoring neuronal function and combating the devastating effects seen in HD.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Türker F, Cook EK, Margolis SS. The proteasome and its role in the nervous system. Cell Chem Biol. 2021;28(7):903–36.33905676 10.1016/j.chembiol.2021.04.003PMC 8286317 · doi ↗ · pubmed ↗
- 2Winckler B, Faundez V, Maday S, et al. The Endolysosomal system and proteostasis: from development to degeneration. J Neurosci. 2018;38(44):9364–9374.30381428 10.1523/JNEUROSCI.1665-18.2018 PMC 6209849 · doi ↗ · pubmed ↗
- 3Liénard C, Pintart A, Bomont P. Neuronal autophagy: regulations and implications in health and disease. Cells. 2024;13(1). doi:10.3390/cells 13010103 PMC 1077836338201307 · doi ↗ · pubmed ↗
- 4Xie Z, Klionsky DJ. Autophagosome formation: core machinery and adaptations. Nat Cell Biol. 2007;9(10):1102–1109.17909521 10.1038/ncb 1007-1102 · doi ↗ · pubmed ↗
- 5Komatsu M, Wang QJ, Holstein GR, et al. Essential role for autophagy protein Atg 7 in the maintenance of axonal homeostasis and the prevention of axonal degeneration. Proc Natl Acad Sci U S A. 2007;104(36):14489–14494.17726112 10.1073/pnas.0701311104 PMC 1964831 · doi ↗ · pubmed ↗
- 6Hara T, Nakamura K, Matsui M, et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 2006;441(7095):885–889.16625204 10.1038/nature 04724 · doi ↗ · pubmed ↗
- 7Komatsu M, Waguri S, Chiba T, et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 2006;441(7095):880–884.16625205 10.1038/nature 04723 · doi ↗ · pubmed ↗
- 8Mizushima N, Yamamoto A, Matsui M, et al. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol Biol Cell. 2003;15(3):1101–1111.14699058 10.1091/mbc.E 03-09-0704 PMC 363084 · doi ↗ · pubmed ↗