Comprehensive evaluation of clinical phenotypes and pathogenic features in late-onset monogenic inflammatory bowel disease: a comparative study with infantile-onset cases
Haoying Liu, Yinxian Shen, Jiaxin Xu, Shangzhan Huang, TingTing Qin, Qi Zhou, Jiazhi Liao, Fang Xiao

TL;DR
This study compares late-onset and infantile-onset monogenic inflammatory bowel disease, finding distinct clinical and genetic differences that could improve diagnosis and treatment.
Contribution
The study provides the first comprehensive comparative analysis of clinical and genetic features between late-onset and infantile-onset monogenic IBD.
Findings
Late-onset mIBD cases show higher rates of Crohn’s disease-like features and intestinal ulcers compared to infantile-onset cases.
Genes like SLCO2A1 and GUCY2C are more commonly implicated in late-onset mIBD, with defects in epithelial and phagocytic functions.
Late-onset cases have fewer perianal disease and oral lesions compared to infantile-onset cases.
Abstract
Monogenic inflammatory bowel disease (mIBD) in patients with onset after the age of 16 has been increasingly recognized, but these reports are sporadic and lack a systematic overview, leaving the clinical and genetic characteristics poorly understood. This study aimed to characterize these late-onset mIBD (LO-mIBD), using an infantile-onset population as a control. Data were extracted from eligible case reports, case series, and cohorts published between January 1990 and October 2023. A comprehensive search of PubMed and Web of Science was conducted following PRISMA guidelines. Demographic, genetic, phenotypic, and genotypic data were collected for the comparative analysis between LO-mIBD (> 16 years) and infantile-onset mIBD (IO-mIBD, < 2 years) cases that met the inclusion criteria. A total of 436 IO-mIBD and 110 LO-mIBD cases were included in the analysis. Patients with LO-mIBD had…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
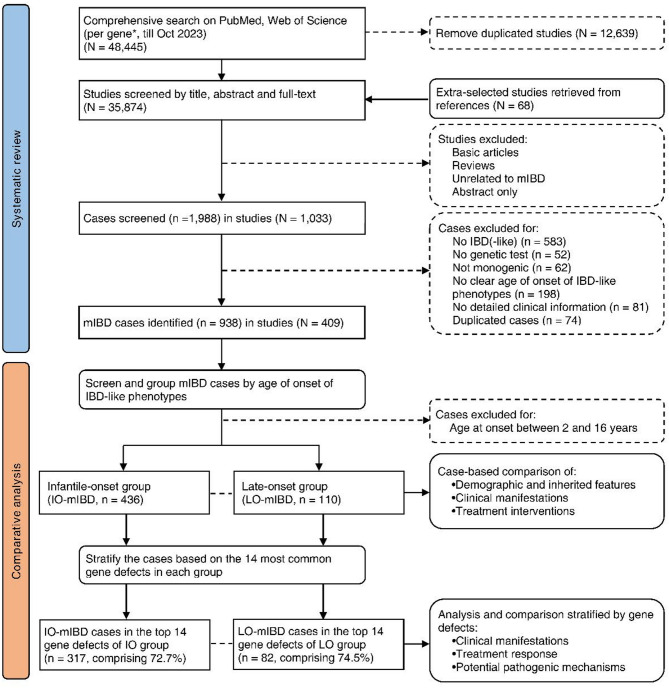 Figure 1
Figure 1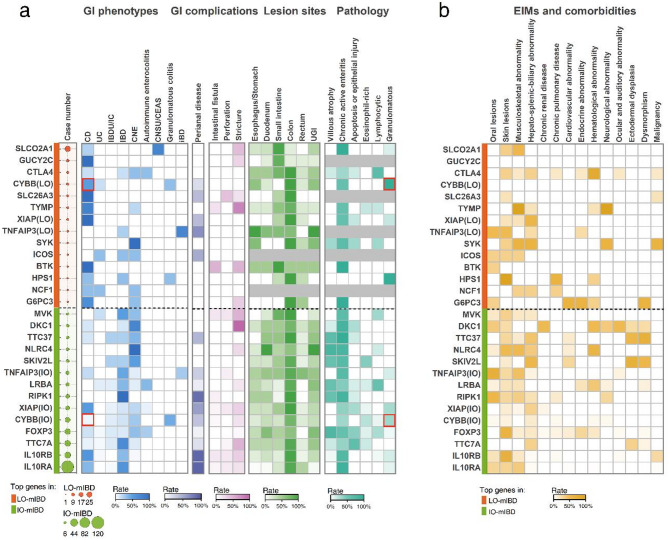 Figure 2
Figure 2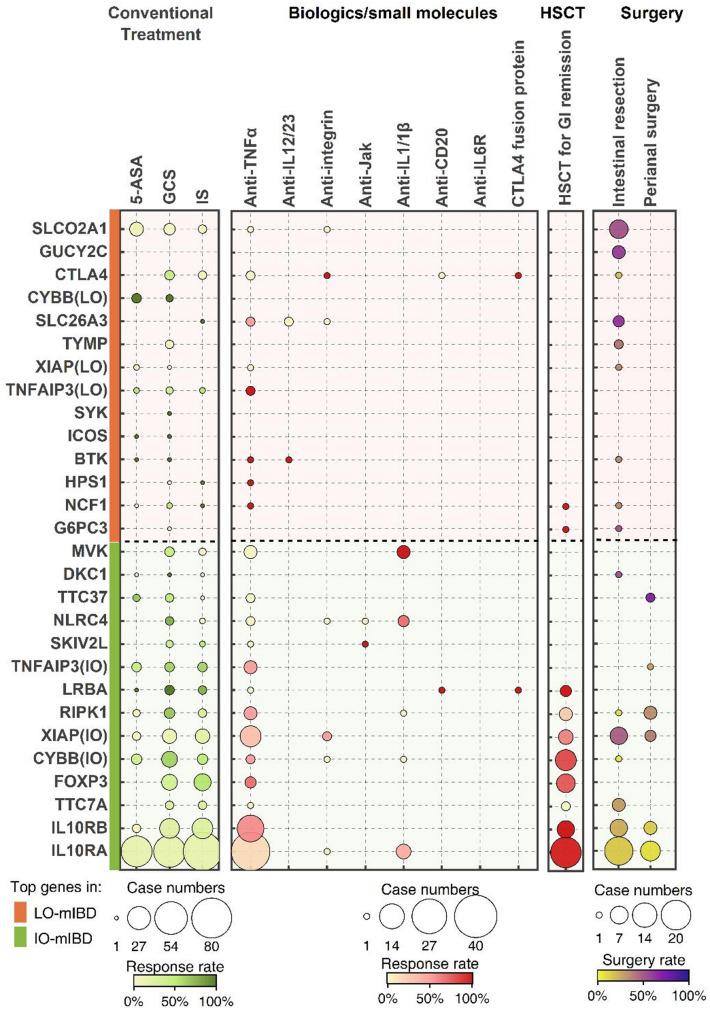 Figure 3
Figure 3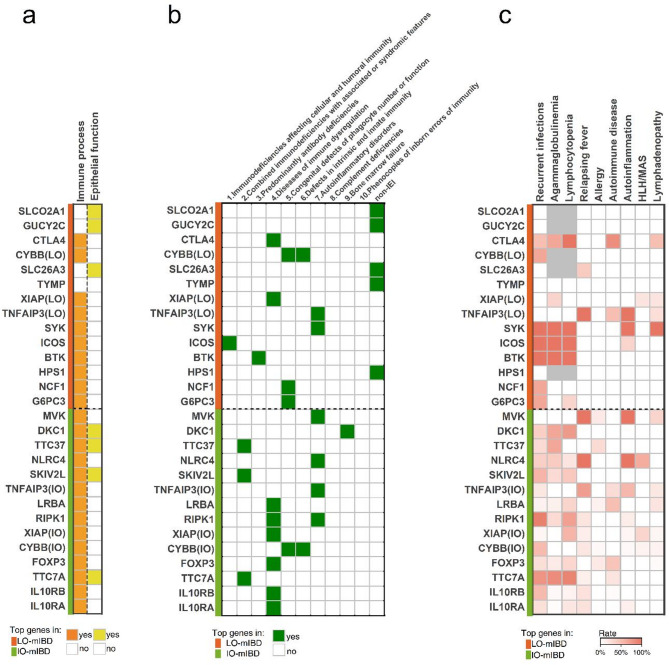 Figure 4
Figure 4- —Subproject of the National Key Research and Development Program
- —https://doi.org/10.13039/501100001809National Natural Science Foundation of China
- —China Crohn’s & Colitis Foundation
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsImmunodeficiency and Autoimmune Disorders · Inflammatory Bowel Disease · Eosinophilic Esophagitis
Introduction
Monogenic inflammatory bowel disease (mIBD) refers to a rare condition characterized by clinical features similar to those of inflammatory bowel disease (IBD), caused by pathogenic mutations in a single gene [1, 2]. These monogenic mutations can lead to various immune or intestinal epithelial dysfunctions, ultimately resulting in intestinal inflammation [3]. Unlike the more prevalent polygenic IBD, such as Crohn’s disease (CD) and ulcerative colitis (UC), mIBD typically follows a more distinct pattern of genetic inheritance that can be diagnosed through genetic sequencing [4, 5]. Additionally, patients with mIBD exhibit various degrees of penetrance and diverse phenotypes in the gastrointestinal (GI) tract, depending on the specific genes involved [6]. For example, patients with interleukin-10 (IL-10) signaling deficiency (mutations in IL10, IL10RA, or IL10RB) present with very early-onset severe intestinal and perianal disease [7], whereas those with FOXP3 deficiency exhibit multisystem autoimmune symptoms and nonspecific enteropathy with villous atrophy [8]. As attention to mIBD grows, the number of reported mIBD cases has considerably increased, as has the discovery of new pathogenic genes [1, 9]. A total of 102 pathogenic genes implicated in mIBD have been identified recently, most of which are associated with primary immunodeficiency disorders (PIDs) or inborn errors of immunity (IEIs) [6, 10].
The GI manifestations of monogenic IBD usually appear at an early age, with patients often classified as the A1 type under the Montreal classification of IBD (before 16 years of age) [11], primarily during infancy or early childhood [9, 12]. The earlier onset of symptoms is one of the key features of mIBD, differentiating mIBD from classic IBD, which commonly emerges during adolescence or early adulthood [13, 14]. However, several patients with monogenic disorders develop IBD-like symptoms beyond childhood [15]. With advances in genetic technology and increased attention to mIBD, many adolescents diagnosed with IBD (Montreal Classification A2/A3, > 16 years) were later identified as having mIBD, especially in cases with atypical features or those refractory to treatment [16–18]. These instances of late-onset mIBD (LO-mIBD) underscore that mIBD is not exclusive to infants, and adult gastroenterologists may encounter such patients, indicating the need to understand the characteristics of the late-onset population. However, LO-mIBD has only been documented in isolated cases (e.g., published late-onset cases with SLCO2A1 [18], SLC26A3 [19], and BTK [20] deficiencies), and to our knowledge, no studies have systematically characterized its clinical and pathophysiological characteristics.
Collectively, LO-mIBD, particularly in individuals with onset after 16 years of age, has historically been overlooked but is closely relevant to adult gastroenterology. To address this gap, we systematically evaluated the clinical features, genotypic characteristics, and pathophysiological mechanisms of LO-mIBD by integrating globally reported cases. In this study, LO-mIBD refers to individuals with mIBD who developed IBD-like phenotypes after the age of 16, corresponding to the A2/A3 type as defined by the Montreal classification [11]. This subgroup was designed as an initial step toward characterizing this understudied population and to ensure clinical translatability in adult gastroenterology practice. In addition, despite existing gaps in LO-mIBD research, the features of infantile-onset mIBD (IO-mIBD, < 2 years) populations have been increasingly elucidated in recent studies [21–23]. Therefore, we conducted a comparative analysis of LO-mIBD using IO-mIBD cases as a control. These efforts are critical for providing initial guidance for adult gastroenterology practices and valuable insights for future research on LO-mIBD.
Methods
Study design and data collection
According to previous studies [6, 9], monogenic IBD was defined as cases with confirmed monogenic disease and IBD-like GI phenotypes. The age at onset of IBD-like manifestations was assessed as the time of endoscopic confirmation or the time at which intestinal symptoms appeared if there was a delay of more than 1 year before endoscopy, as described by Bolton et al. [6].
A comprehensive search was conducted for articles published between January 1990 and October 2023 using PubMed and Web of Science, according to the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) guidelines (Fig. 1). The search terms included “IBD”, “inflammatory bowel disease”, “Crohn’s”, “enterocolitis”, and “enteropathy”, combined with the names or abbreviations of genes, proteins, monogenic diseases, or syndromes. A search was performed for each of the 102 defined mIBD genes [2, 6]. Google Scholar, the related article function, and reference lists were also used to supplement the published studies; no language restrictions were made. The titles, abstracts, and full texts were evaluated to exclude basic scientific articles and reviews without clinical cases, articles unrelated to mIBD, conference abstracts, and duplicate articles. Case details were further scrutinized to screen for IO-mIBD and LO-mIBD during the full-text evaluation of the eligible articles. The inclusion criteria for the cases were as follows: (1) definite diagnosis of monogenic disease and corresponding pathogenic single-gene mutations confirmed by genetic sequencing, including Sanger sequencing, targeted gene panel sequencing (TGPS), targeted next-generation sequencing (TNGS), whole-exome sequencing (WES), and whole-genome sequencing (WGS); (2) presentation of IBD-like GI phenotypes or chronic intestinal inflammation based on endoscopic or histological findings, including suspected diagnoses of IBD, intestinal Behçet’s disease, granulomatous colitis, autoimmune enteropathy, chronic nonspecific enterocolitis, cryptogenic multifocal ulcerous stenosing enteritis (CMUSE), chronic non-specific multiple ulcers of the small intestine (CNSU), and common variable immunodeficiency-associated enteropathy; (3) the age at onset of IBD-like manifestations was either younger than 2 years or older than 16 years; and (4) detailed clinical information for individual cases, including at least two of the following four aspects: demographic information, GI findings, extraintestinal manifestations (EIMs), and treatment information. The exclusion criteria were as follows: (1) no genetic testing results or no genetic testing performed; (2) mutations in multiple genes; (3) no evidence of intestinal inflammation, or enterocolitis caused by other known factors, such as pathogens; (4) IBD-like manifestations that appeared between 2 and 16 years of age; and (5) insufficient details of individual cases in cohort studies or case series.
The following data were collected for each case: pathogenic genes, mutation zygosity, age at onset of IBD-like disease, sex, parental consanguinity, family history, GI phenotypes labeled by the authors, GI symptoms and complications, lesion sites, endoscopic and pathological features, EIMs and comorbidities, and treatment interventions and responses. See the Supporting Information (Supplementary Table 1) for the detailed classification and definition of each feature. Briefly, the mutation zygosities included homozygotes, compound heterozygotes, heterozygotes, and hemizygotes. GI phenotypes were classified into five categories: CD, UC, IBD-unclassified (IBDU) or indeterminate colitis (IC), IBD (IBD cases without delineating subtypes), and others (other chronic intestinal inflammatory diseases resembling IBD). Intestinal pathological features were classified into five patterns, as described by Wilkins et al. [24]. EIMs are assessed based on the systemic manifestations associated with IBD [25], along with other specific comorbidities, such as recurrent infections, fever, malignancy, etc. Treatment interventions include traditional options for classic IBD and several other special approaches for mIBD, such as hematopoietic stem cell transplantation (HSCT). Response to therapy refers to symptomatic or endoscopic remission of the GI tract, excluding transient relief. The variants in the included cases were re-examined using the classification tool “Franklin” (https://franklin.genoox.com/clinical-db/home) based on the latest ACMG guidelines [26]. The quality of the included cases was assessed using the Joanna Briggs Institute (JBI) critical appraisal checklists for case reports and case series (Supplementary Table 2A, B). Each item was scored as 1 (“yes”) or 0 (“no/unclear”), with maximum total scores of 8 and 10, respectively. Studies scoring > 7, 4–6, and 0–3 were considered high, moderate, and low quality, respectively. Two authors independently evaluated the procedure.
We comparatively analyzed the demographic, inherited, and clinical data of the patients with LO-mIBD and IO-mIBD. The 14 most common genes were used for further stratification to investigate the genotype-associated phenotypic features and treatment effectiveness in those with LO-mIBD and IO-mIBD. Finally, the immune process was evaluated according to the latest IEIs/PIDs classification [10], and Gene Ontology (GO) enrichment was analyzed using Metascape [27] to compare the functional pathways and underlying pathophysiological mechanisms of the most common genes in each group.
Statistical analysis
Median and interquartile range (IQR) were used to describe the age at onset, whereas numbers and percentages were used for other categorical variables. The chi-square test or Fisher’s exact test was used to compare rates or proportions, and the Bonferroni method was used for p-value adjustment in post hoc multiple comparisons. Univariate binary logistic regression was used to analyze the correlation between variant zygosity and LO-mIBD, and multifactorial logistic regression was used to adjust potential confounders, including sex, consanguineous history, GI complications, and genotype. The results are presented as the odds ratio (OR) of LO-mIBD relative to IO-mIBD, along with its 95% confidence interval (CI). Sensitivity analysis was conducted to evaluate the potential risk of bias arising from missing data, assuming an RI_LTFU/FU_ of 0.5 for the group with the higher incidence and 1 for the group with the lower incidence [28]. P values less than 0.05 were considered statistically significant. All statistical analyses were conducted using the SPSS software (v25.0; IBM, New York, USA), and figures were generated using GraphPad Prism (v10.1) and Origin (v2025).
Results
Demographic and inherited features of patients with late-onset and infantile-onset mIBD
We initially screened 1,988 cases from 1,033 eligible articles by searching for 102 defined mIBD genes. According to our criteria, a total of 938 mIBD cases involving 88 of 102 genes were discovered, among which 546 cases involving 77 genes were ultimately included in the comparative analysis (Fig. 1).
Specifically, 436 cases of IO-mIBD and 110 cases of LO-mIBD were included, with males accounting for 61.7% and 48.1% of the cases, respectively (p = 0.011). The median age at the onset of the IBD-like disease was 0.16 years (IQR: 0.04–0.50) in patients with IO-mIBD and 24.00 years (IQR: 18.00–35.25) in those with LO-mIBD. Asians comprised the largest proportion of included cases (36.45%), followed by European/Caucasians (12.45%), with race/ethnicity unreported in 34.98% of cases (Supplementary Fig. 3b). Parental consanguinity, referring to the offspring of consanguineous parents, was less prevalent in patients with LO-mIBD than in those with IO-mIBD (p = 0.015, OR: 0.49, 95% CI: 0.27–0.88), and it was mainly found in patients with homozygous mutations (100% in LO-mIBD and 98.1% in IO-mIBD). No significant differences in the family history of either monogenic disorders or IBD-like manifestations were observed between the groups (p > 0.05).
Homozygosity was the most common type of zygosity in both groups. However, patients with LO-mIBD had a larger proportion of heterozygotes and a lower proportion of compound heterozygotes compared with IO-mIBD cases (Bonferroni-adjusted p < 0.05, Table 1). Moreover, the results of binary logistic regression analysis revealed a significant correlation between the zygosity type of heterozygosity and LO-mIBD (p = 0.001, OR: 2.56, 95% CI: 1.45–4.52, using homozygotes as the reference, Supplementary Table 3). Multifactorial regression analyses adjusted for genotype, rather than sex or consanguineous history, significantly altered the differences in zygosity, suggesting that genotype is a key factor influencing the differences in zygosity between the groups (Supplementary Table 4). Further analysis showed that the proportion of patients with autosomal dominant gene defects was also higher in the LO-mIBD group (23.6% vs. 10.8%, adjusted p < 0.05), with GUCY2C, CTLA4, and SYK deficiencies being the most common.
A total of 65 of the 77 pathogenic genes were reported in patients with IO-mIBD, 36 in patients with LO-mIBD, and 24 overlapped between the groups (Supplementary Fig. 1). The median age of onset of IBD-like phenotypes varied across different genes, ranging from 0.02 to 27 years. Specifically, 33 genes had a median age at onset of less than 2 years, whereas 9 had an onset age greater than 16 years, including CARD8, PIK3R1, ICOS, PI4KA, TRNT1, TYMP, IRF2BP2, GUCY2C, and SYK (Supplementary Fig. 2).
Fig. 1PRISMA flow chart of the study design
The comprehensive search and screening are based on PRISMA guidelines. A total of 436 cases of infantile-onset mIBD (IO-mIBD) and 110 cases of late-onset mIBD (LO-mIBD) meeting the criteria were included in the comparative analysis. The cases were then stratified according to the top 14 most common gene defects in each group to analyze the clinical and pathogenic characteristics associated with these defects.
^*^. A total of 102 mIBD genes identified by Bolton et al. [6] and Kammermeier et al. [2].
Table 1. Comparison of demographic and inherited features in patients with late-onset vs. infantile-onset mIBDLO-mIBDIO-mIBDOR (95% CI)P value% (n/N^†^)% (n/N^†^) Total number of cases 110436Onset age of IBD (years)(median, IQR)24.00(18.00, 35.25)0.16(0.04, 0.50) Sex (male) 48.1 (52/108)61.7 (265/428)0.57 (0.38, 0.88)0.011 Parental consanguinity 17.4 (16/92)30.1 (107/356)0.49 (0.27, 0.88)0.015 Family history of similar monogenic disorders 45.7 (42/92)36.5 (130/356)1.46 (0.92, 2.32)0.108 Family history of IBD-like manifestations 28.3 (26/92)31.5 (112/356)0.86 (0.52, 1.42)0.553 Zygosity < 0.001 Homozygotes40.9 (45/110)42.0 (183/436)(Ref)(Ref) Compound heterozygotes19.1 (21/110) ^^28.7 (125/436)0.68 (0.39, 1.20)0.187 Heterozygotes26.4 (29/110) ^^10.6 (46/436)2.56 (1.45, 4.52)0.001 Hemizygotes13.6 (15/110)18.8 (82/436)0.74 (0.392, 1.41)0.365IQR: interquartile range^†^. n = number of cases presenting the corresponding features; N = number of cases with available information about the features in the reports^**^. Bonferroni-adjusted P < 0.05 between LO-mIBD and IO-mIBD
Clinical manifestations of late-onset mIBD compared with infantile-onset mIBD
Among the five categories of GI phenotypes analyzed, the CD-like phenotype was the most common in LO-mIBD (43.6%), followed by other intestinal inflammatory diseases, particularly chronic nonspecific multiple ulcers of the small intestine (CNSU) and chronic enteropathy associated with the SLCO2A1 gene (CEAS) (17.3%). The prevalence of these GI phenotypes in the LO-mIBD group was significantly higher than that in the IO-mIBD group (adjusted p < 0.05, Table 2), which was more commonly labeled as IBD without delineating subtypes (adjusted p < 0.05).
Patients with LO-mIBD presented significantly more incidences of abdominal pain (p < 0.001, OR: 4.62, 95% CI: 2.80–7.62) but fewer diarrhea, hematochezia, and perianal diseases compared with IO-mIBD cases (p < 0.001, OR: 0.16, 95% CI: 0.09–0.29; OR: 0.29, 95% CI: 0.18–0.48; OR: 0.10, 95% CI: 0.05–0.20, respectively). GI complication of stricture was commonly reported in patients with LO-mIBD (31.5%), markedly higher than in those with IO-mIBD (p = 0.002, OR: 2.24, 95% CI: 1.32–3.81). Complications such as intestinal fistula and perforation were similar between the groups (p > 0.05). The colon was the most common site of lesions in both groups. However, small intestinal lesions were relatively more common in patients with LO-mIBD than in those with IO-mIBD (p < 0.001, OR: 3.37, 95% CI: 2.08–5.45), while colonic (p < 0.001, OR: 0.25, 95% CI: 0.15–0.43) and rectal lesions (p = 0.021, OR: 0.45, 95% CI: 0.22–0.90) showed the opposite. Patients with LO-mIBD also had higher incidences of intestinal ulcers (p = 0.008, OR: 3.21, 95% CI: 1.31–7.86) but fewer pseudopolyps (p = 0.006, OR: 0.21, 95% CI: 0.06–0.70) and villous atrophy (p = 0.005, OR: 0.20, 95% CI: 0.06–0.68) compared with IO-mIBD cases. Among the five pathological patterns, the classic pattern—chronic active enteritis—was most prevalent in both groups. The lymphocytic pattern, characterized by lymphocytosis and nodular lymphoid hyperplasia, as well as the granulomatous pattern, were significantly more common in patients with LO-mIBD than in those with IO-mIBD (p = 0.024, OR: 2.36, 95% CI: 1.10–5.05; p = 0.025, OR: 2.43, 95% CI: 1.10–5.38, respectively).
Table 3 shows the EIMs and comorbidities reported by patients with LO-mIBD and IO-mIBD. Notably, those with LO-mIBD had significantly higher instances of anemia and musculoskeletal abnormalities than those with IO-mIBD (p = 0.004, OR: 1.98, 95% CI: 1.23–3.18; p < 0.001, OR: 3.45, 95% CI: 2.06–5.77, respectively), whereas growth retardation (p = 0.001, OR: 0.16, 95% CI: 0.09–0.30), oral lesions (p = 0.005, OR: 0.39, 95% CI: 0.20–0.77), recurrent infections (p = 0.024, OR: 0.60, 95% CI: 0.38–0.94), and fever (p = 0.001, OR: 0.31, 95% CI: 0.15–0.63) were less common. The number of EIMs reported by the patients did not significantly differ between the groups (p > 0.05). CEAS/CNSU caused by SLCO2A1 defects, as well as congenital/familial diarrhea associated with SLC26A1, SLC9A3, and GUCY2C mutations, were the most prevalent monogenic disorders in patients with LO-mIBD, significantly more common than in those with IO-mIBD (p < 0.001, Table 4). In contrast, IL-10 signaling deficiency was notably prevalent in patients with IO-mIBD, with no case reports in those with LO-mIBD (p < 0.001).
Multifactorial logistic regression analysis was conducted to evaluate the influence of confounding factors (Supplementary Table 4). The results remained statistically significant after adjusting for age and parental consanguinity. Adjustment for genotype (analysis based on cases within the 24 overlapping genes) altered the significance of features such as zygosity, GI complication of stricture, lesions in the duodenum and rectum, erosions, pseudopolyps, villous atrophy, and oral EIMs. This suggests that genotype plays a critical role in driving differences in these phenotypic features.
The percentage of missing data between the groups did not significantly differ in the above analysis (2.9–51.8% and 0–50% in the IO-mIBD and LO-mIBD groups, respectively, p > 0.05). The results of sensitivity analysis (Supplementary Table 6) revealed that the differences remained significant when the analysis was conducted using the assumed RI_LTFU/FU_, except for ulcers and pathological features, for which approximately 50% of the data were missing. Approximately 60–80% of the included cases were derived from high-quality case reports and case series (Supplementary Fig. 3a). WES (43.59%) was the sequencing technology most frequently used for diagnosis, followed by Sanger sequencing (28.2%) and TGPS/TNGS (16.3%, Supplementary Fig. 3c). The reevaluation of variant pathogenicity according to the latest ACMG guidelines showed that over 90% of the included cases had pathogenic and likely pathogenic variants (76.19% and 16.85%, respectively, Supplementary Fig. 3d). No significant differences were found in the genetic testing technologies or variant pathogenicity between the groups. These findings collectively suggest a low risk of bias from missing data and limited confounding effects resulting from the heterogeneity of the reports and genetic testing technologies.
Table 2. Comparison of gastrointestinal manifestations in patients with late-onset vs. infantile-onset mIBDLO-mIBDIO-mIBDOR (95% CI)P value% (n/N^†^)% (n/N^†^) GI phenotypes < 0.001 CD43.6 (48/110) ^^24.1 (105/436)8.53 (4.02, 18.11)< 0.001 UC5.5 (6/110)4.1 (18/436)6.22 (1.99, 19.49)0.002 IBDU/IC0.9 (1/110)3.9 (17/436)1.10 (0.13, 9.20)0.931 IBD^‡^8.2 (9/110) ^^38.5 (168/436)(Ref)(Ref) Others^§^41.8 (46/110) ^****^29.4 (128/436)6.71 (3.17, 14.21)< 0.001 GI symptoms Abdominal pain46.2 (43/93)15.7 (54/344)4.62 (2.80, 7.62)< 0.001 Diarrhea66.7 (62/93)92.4 (318/344)0.16 (0.09, 0.29)< 0.001 Hematochezia31.2 (29/93)60.8 (209/344)0.29 (0.18, 0.48)< 0.001 Perianal disease10.6 (10/94)53.7 (202/376)0.10 (0.05, 0.20)< 0.001 GI complications Intestinal fistula2.2 (2/89)5.7 (19/335)0.38 (0.09, 1.67)0.294 Perforation4.5 (4/89)7.5 (25/335)0.58 (0.20, 1.72)0.324 Stricture31.5 (28/89)17.0 (57/335)2.24 (1.32, 3.81)0.002 Lesion sites Esophagus/Stomach23.6 (21/89)19.0 (65/343)1.32 (0.76, 2.31)0.328 Duodenum20.2 (18/89)25.1 (86/343)0.76 (0.43, 1.34)0.340 Small intestine58.4 (52/89)29.4 (101/343)3.37 (2.08, 5.45)< 0.001 Colon62.9 (56/89)87.2 (299/343)0.25 (0.15, 0.43)< 0.001 Rectum11.2 (10/89)22.2 (76/343)0.45 (0.22, 0.90)0.021 Upper GI involvement^¶^31.5 (28/89)31.2 (107/343)1.01 (0.61, 1.67)0.962 Endoscopic findings Ulcer90.9 (60/66)75.7 (159/210)3.21 (1.31, 7.86)0.008 Erosion18.2 (12/66)22.9 (48/210)0.75 (0.37, 1.52)0.422 Pseudopolyps4.5 (3/66)18.6 (39/210)0.21 (0.06,0.70)0.006 Pathological features Villous atrophy5.5 (3/55)22.9 (52/236)0.20 (0.06, 0.68)0.005 Chronic active enteritis63.6 (35/55)71.2 (168/236)0.71 (0.38, 1.31)0.272 Apoptosis or epithelial injury7.3 (4/55)13.6 (32/236)0.50 (0.17, 1.48)0.202 Eosinophil-rich7.3 (4/55)8.1 (19/236)0.90 (0.29, 2.75)1.000 Lymphocytic20.0 (11/55)9.3 (22/236)2.43 (1.10, 5.38)0.025 Granulomatous21.8 (12/55)10.6 (25/236)2.36 (1.10, 5.05)0.024CD, Crohn’s disease; UC, ulcerative colitis; IBDU, IBD-unclassified; IC, indeterminate colitis; GI, gastrointestinal tract^†^. n = number of cases presenting the corresponding features; N = number of cases with available information about the features in the reports^‡^. Gastrointestinal phenotypes labeled as IBD without specifying the exact subtype^§^. Other chronic intestinal inflammation resembling IBD, see detailed definitions in Supplementary Table 1B^¶^. The upper gastrointestinal tract includes the esophagus, stomach, and duodenum^^. Bonferroni-adjusted P < 0.05 between LO-mIBD and IO-mIBD
Table 3. Comparison of extraintestinal manifestations and comorbidities in patients with late-onset vs. infantile-onset mIBDLO-mIBDIO-mIBDOR (95% CI)P value% (n/N^†^)% (n/N^†^) Extraintestinal manifestations >1 EIMs49.1 (54/110)51.3 (217/423)0.92 (0.60, 1.39)0.680 >2 EIMs29.1 (32/110)26.5 (112/423)1.14 (0.72, 1.81)0.582 Anemia30.9 (34/110)18.4 (78/423)1.98 (1.23, 3.18)0.004 Growth retardation11.8 (13/110)44.9 (190/423)0.16 (0.09, 0.30)< 0.001 Oral lesions10.0 (11/110)22.0 (93/423)0.39 (0.20, 0.77)0.005 Skin lesions29.1 (32/110)36.9 (156/423)0.70 (0.45, 1.11)0.128 Musculoskeletal abnormality29.1 (32/110)10.6 (45/423)3.45 (2.06, 5.77)< 0.001 Hepato-splenic-biliary abnormality13.6 (15/110)13.9 (59/423)0.97 (0.53, 1.79)0.933 Chronic renal disease1.8 (2/110)1.9 (8/423)0.96 (0.20, 4.59)1.000 Chronic pulmonary disease7.3 (8110)3.3 (14/423)2.29 (0.94, 5.61)0.111 Cardiovascular abnormality3.6 (4/110)4.3 (18/423)0.85 (0.28, 2.56)0.983 Endocrine abnormality5.5 (6/110)4.0 (17/423)1.38 (0.53, 3.58)0.692 Hematological abnormality10.9 (12/110)7.6 (32/423)1.50 (0.74, 3.01)0.256 Neurological abnormality9.1 (10/110)5.4 (23/423)1.74 (0.80, 3.77)0.157 Ocular and auditory abnormality1.8 (2/110)2.8 (12/423)0.63 (0.14, 2.88)0.794 Lymphadenopathy7.3 (8/110)4.0 (17/423)1.87 (0.79, 4.46)0.150 Comorbidities Recurrent infections30.0 (33/110)41.8 (177/423)0.60 (0.38, 0.94)0.024 Relapsing fever8.2 (9/110)22.5 (95/423)0.31 (0.15, 0.63)0.001 Autoimmune disease10.9 (12/110)9.0 (38/423)1.24 (0.63, 2.46)0.537 Autoinflammation8.2 (9/110)10.9 (46/423)0.73 (0.35, 1.54)0.408 HLH/MAS0.9 (1/110)3.3 (14/423)0.27 (0.04, 2.06)0.302 Allergy0.9 (1/110)2.4 (10/423)0.38 (0.05, 2.99)0.562 Ectodermal dysplasia3.6 (4/110)5.7 (24/423)0.63 (0.21, 1.85)0.394 Dysmorphism3.6 (4/110)6.4 (27/423)0.55 (0.19, 1.62)0.273 Malignancy5.5 (6/110)3.8 (16/423)1.47 (0.56, 3.84)0.606^†^. n = number of cases presenting the corresponding features; N = number of cases with available information about the features in the reportsEIMs, extraintestinal manifestations; HLH, hemophagocytic lymphohistiocytosis; MAS, macrophage activation syndrome
Table 4. Prevalence of monogenic disorders in patients with late-onset and infantile-onset mIBDMonogenic disordersPathogenic genesLO-mIBDn (%)N = 110IO-mIBDn (%)N = 436P valueIL-10 signaling deficiencyIL10,* IL10RA*,* IL10RB0 (0.0)156 (35.8)< 0.001IPEX or IPEX-likeIL2RA*,* MALT1*,* FOXP3*,* BACH2*,* LRBA*,* CTLA4*,* STAT1*,* STAT3*,* CARD11*,* ITCH11 (10.0)55 (12.6)0.516CGDCYBB*,* CYBA*,* CYBC1*,* NCF1*,* NCF2*,* NCF411 (10.0)27 (6.2)0.206CEAS/CNSU SLCO2A1 26 (23.6)5 (1.2)< 0.001XLP2 XIAP 5 (4.6)15 (3.4)0.789SD/THESSKIV2L*,* TTC370 (0.0)16 (3.67)0.085Congenital/familial diarrheaSLC26A3*,* SLC9A3*,* GUCY2C12 (10.9)4 (0.9)< 0.001HA20 TNFAIP3 4 (3.6)11 (2.5)0.755WAS or WAS-likeWAS*,* ARPC1B*1 (0.9)10 (2.3)0.587Only monogenic disorders or syndromes with a total of more than 10 included cases were listed. Abbreviations: IL-10: interleukin-10; IPEX: X-linked immune dysregulation, polyendocrinopathy, enteropathy syndrome; CGD, chronic granulomatous disease; CEAS, chronic enteropathy associated with the SLCO2A1 gene; CNSU, chronic nonspecific multiple ulcers of the small intestine; XLP2, X-linked lymphoproliferative syndrome 2; SD/THES, syndromic diarrhea/tricho-hepato-enteric syndrome; HA20, haploinsufficiency of A20; WAS, Wiskott–Aldrich syndrome;
Genotype-associated clinical manifestations and treatment of late-onset and infantile-onset mIBD
Given the heterogeneity and phenotype-genotype associations of mIBD, it is necessary to investigate the clinical manifestations from the perspective of genotypes to further understand the phenotypic characteristics of LO-mIBD. Due to the complex genotypes and dispersed cases (Supplementary Fig. 4), we focused on the top 14 common pathogenic genes in each group, which accounted for the majority of cases (72.21% and 74.55% in the IO-mIBD and LO-mIBD groups, respectively). Three genes—CYBB, XIAP, and TNFAIP3—overlapped between the groups.
Figure 2 shows the clinical manifestations stratified by the 14 common genes in each group. Consistent with the above observations, the GI lesions in LO-mIBD with various monogenetic deficiencies were predominantly characterized by a CD-like phenotype, particularly in cases caused by GUCY2C, SLC26A3, XIAP, BTK, TYMP, G6PC3, and CYBB mutations, with a proportion of no less than 50%. In contrast, IBD and chronic nonspecific enterocolitis (CNE) were the main phenotypes associated with several common monogenic defects in those with IO-mIBD. In addition to the CD-like phenotype, patients with LO-mIBD caused by ICOS deficiency could be diagnosed with UC, IBD, and CNE; those with TNFAIP3 deficiency could be diagnosed with IBD and intestinal Behçet’s disease (iBD); and those with SYK mutations were characterized by CNE. Intestinal strictures were notably prevalent in patients with LO-mIBD, particularly in those caused by TYMP, BTK, SLCO2A1, G6PC3, GUCY2C, and SLC26A3 mutations. The EIMs and comorbidities in IO-mIBD and LO-mIBD were found to be complex and multi-systemic, depending on the specific genes involved (Fig. 2b).
Regarding the three genes that were common in both groups, CYBB deficiency usually leads to granulomatous colitis with perianal disease, which is often diagnosed as CD or chronic granulomatous disease (CGD). However, the initial diagnosis of CD (50.0% vs. 4.8%, p < 0.05) and pathological findings of granulomas (100.0% vs. 33.3%, p = 0.029) were more prominent in patients with LO-mIBD than in those with IO-mIBD. TNFAIP3 deficiency typically causes intestinal, oral, and genital ulcers, which are frequently diagnosed as iBD or CD. Patients with LO-mIBD were more likely to be diagnosed with iBD than those with IO-mIBD (75.0% vs. 27.3%), although this difference was not statistically significant (p > 0.05). XIAP deficiency is typically characterized by CD-like enteropathy and perianal disease.
The treatment choices differed somewhat between patients with IO-mIBD and LO-mIBD (Supplementary Table 7). Surgical resection was significantly more prevalent in patients with LO-mIBD, particularly in those caused by SLC26A3, GUCY2C, SLCO2A1, and G6PC3 mutations, with a resection rate of approximately 50.0%. The resection rates remained statistically significant after adjusting for GI complications or genotypes in the multifactorial regression analysis (Supplementary Table 5). HSCT was performed in 26.5% of IO-mIBD cases, providing intestinal benefits for those with certain monogenic defects, such as IL10RA/B, CYBB, XIAP, and LRBA deficiencies. However, only three cases of LO-mIBD—with G6PC3, NCF1, and LRBA mutations, respectively—underwent HSCT and showed GI improvement. These results are exploratory due to the small sample size and need to be confirmed by future studies. The response to traditional medications in patients with mIBD varied primarily according to the underlying monogenic defects (Fig. 3). However, among patients with CYBB deficiency, those with LO-mIBD had higher response rates to 5-aminosalicylic acid (5-ASA, 100.0% vs. 33.3%) and glucocorticoids (GCS, 100.0% vs. 66.7%) than those with IO-mIBD, although the difference did not reach significance (p > 0.05). Some other treatments displayed GI effectiveness in certain monogenic defects with limited case numbers, such as anti-TNFα biologics for patients with LO-mIBD caused by BTK, NCF1, and TNFAIP3 deficiencies; anti-IL12/23 biologics for those caused by BTK deficiency; CTLA4 fusion protein (abatacept) for those caused by LRBA and CTLA4 deficiencies; and anti-IL-1β/1R biologics for patients caused by mutations of IL10RA, NLRC4, and MVK, etc.
Fig. 2. Clinical manifestations in patients with late-onset and infantile-onset mIBD stratified by the 14 most common gene defects. (a) The number of cases and GI manifestations stratified by gene defects. Red and green bars denote the 14 most prevalent pathogenic genes in patients with LO-mIBD and IO-mIBD, respectively. Dot size indicates the number of cases within each gene, with red and green dots representing the LO-mIBD and IO-mIBD cases, respectively. The color intensity of the square represents the incidence of the corresponding features, from 0% to 100%, whereas gray indicates unavailable data. The red border indicates a significant difference between patients with IO-mIBD and LO-mIBD caused by the same gene defects (p < 0.05). (b) Extraintestinal manifestations and comorbidities stratified by gene defects. The color intensity of the square represents the incidence of the corresponding features, ranging from 0% to 100%. Abbreviations: CD, Crohn’s disease; UC, ulcerative colitis; IBDU, IBD-unclassified; IC, indeterminate colitis; IBD, refers to the category of IBD without delineating specific subtypes in this study; UGI: upper gastrointestinal tract, mainly in anatomical terms; CNE, chronic nonspecific enterocolitis; CNSU, chronic nonspecific multiple ulcers of the small intestine; CEAS, chronic enteropathy associated with the SLCO2A1 gene; iBD, intestinal Behçet’s disease; EIMs, extraintestinal manifestations
Fig. 3. Treatment interventions and responses in late-onset and infantile-onset mIBD stratified by gene defects. Stratification of the therapeutic response to conventional treatment, biologics/small molecules, and HSCT, as well as the surgical rates in patients with IO-mIBD and LO-mIBD according to the 14 most common gene defects. Red and green bars denote the 14 most prevalent pathogenic genes in patients with LO-mIBD and IO-mIBD, respectively. The dot size represents the number of patients receiving treatment. The color intensity indicates the response rate to treatment, ranging from 0% to 100%. Abbreviations: 5-ASA, 5-aminosalicylic acid; GCS, glucocorticoids; IS, immune suppressants and modulators, including azathioprine, methotrexate, cyclosporine, thalidomide, tacrolimus, and sirolimus; HSCT, hematopoietic stem cell transplantation
Gene function and intestinal pathogenesis underlying monogenetic defects in late-onset mIBD compared with infantile-onset mIBD
The age of onset of IBD-like phenotypes varies with the gene, suggesting an association between the pathogenesis of gene defects and the age of onset. It remains unclear whether the pathogenic mechanisms of genetic defects between IO-mIBD and LO-mIBD are similar or distinct. The GI pathogenesis of mIBD primarily involves defects in immune function and epithelial maintenance. We thus compared the pathogenic mechanisms and immune-related manifestations based on the 14 most common gene defects in each group.
All of the 14 most common genes in the IO-mIBD group were functionally related to the immune process (Fig. 4a), and their deficiencies are included in the latest IEI classification, with “diseases of immune dysregulation” being the most common IEIs/PIDs category (Fig. 4b). Patients with IO-mIBD caused by these gene defects usually exhibited immune abnormalities such as recurrent infections, agammaglobulinemia, lymphocytopenia, autoimmune disease, and autoinflammation (Fig. 4c). In addition, genes such as TTC7A, SKIV2L, TTC37, and DKC1 also play a role in epithelial maintenance, with their defects often leading to ectodermal dysplasia, dysmorphism, and intestinal epithelial damage (Fig. 2b).
For the top 14 gene defects in the LO-mIBD group, the most common IEIs/PIDs category was “congenital defects of phagocyte number or function”, involving G6PC3, CYBB, and NCF1 deficiencies (Fig. 4b). In addition, several gene defects have not been included in the IEIs/PIDs classification, including SLCO2A1, GUCY2C, SLC26A3, TYMP, and HPS1 mutations. The proportion of non-IEI gene defects was significantly higher in the LO-mIBD group than in the IO-mIBD group (p < 0.05). Patients with LO-mIBD caused by these gene mutations rarely present with immune abnormalities (Fig. 4c). Furthermore, these non-IEI gene defects can be pathogenic via nonimmune processes, particularly epithelial dysfunction. For example, SLC26A3 and GUCY2C mutations impair epithelial ion transport, while SLCO2A1 mutations disrupt prostaglandin transport and compromise the intestinal barrier (Supplementary Table 8). The results of GO enrichment analysis of the common genes specific to each group further supported these findings. GO terms associated with immune and inflammatory regulation were significantly enriched in the IO-mIBD group, whereas those related to the response to pathogens and ion transport were enriched in the LO-mIBD group (p < 0.05; Supplementary Table 9).
Fig. 4. Pathogenesis and immune-associated abnormalities of common gene defects in patients with late-onset and infantile-onset mIBD. (a) The involvement of the immune process and epithelial function of the top 14 common genes in patients with IO-mIBD and LO-mIBD, respectively. The blank boxes indicate no, whereas the orange and yellow boxes indicate yes. (b) The categories of inborn errors of immunity (IEI) classification of gene defects, according to the 2022 classification from the International Union of Immunological Societies (IUIS). Non-IEI refers to genes that are not included in the IEI classification. The blank and green boxes indicate no and yes, respectively. (c) The incidence of immune-related abnormalities stratified by gene defects. The color intensity represents the incidence ranging from 0% to 100%, with gray indicating unavailable data. Abbreviations: HLH, hemophagocytic lymphohistiocytosis; MAS, macrophage activation syndrome
Discussion
LO-mIBD remains underexplored despite its increasing relevance in clinical practice, particularly in adult gastroenterology. This study provides the first comprehensive evaluation of the LO-mIBD population with IBD-like phenotypes developing after 16 years of age, revealing some distinct clinical and genetic features compared with IO-mIBD cases. Our findings bridge the knowledge gaps regarding LO-mIBD and provide novel insights to facilitate clinical management and future research.
The genetic landscape of mIBD follows Mendelian inheritance patterns, as reflected in family history, consanguinity, zygosity types, and inheritance modes [1, 9]. Both patients with LO-mIBD and IO-mIBD had high rates of positive family history and were predominantly affected by homozygous and autosomal recessive mutations, consistent with the findings of Nambu et al. [9]. However, patients with LO-mIBD had less parental consanguinity and more heterozygous mutations in autosomal dominant genes (e.g., GUCY2C, CTLA4, SYK) than those with IO-mIBD. These findings highlight the importance of genetic counseling for patients with LO-mIBD, as autosomal dominant mutations confer a 50% risk of transmission to their offspring.
The GI phenotypes of patients with mIBD are highly heterogeneous [1]. A previous study of all published cases of mIBD has revealed that most cases were classified as IBD without specifying the subtype, followed by CD [9]. However, data specifically on late-onset populations are lacking. Our study revealed that the CD-like phenotype was the most common presentation in patients with LO-mIBD and was more prevalent than in those with IO-mIBD. These findings are consistent with those of Crowley et al. [23], who reported CD-like phenotypes in the majority of late-onset mIBD cases diagnosed during late childhood and early adolescence. Importantly, our findings confirm and extend the previous results of CD-like phenotypes in late-onset mIBD populations by showing higher rates of intestinal ulcers, strictures, small bowel lesions, and granulomas in LO-mIBD.
The clinical manifestations of patients with LO-mIBD are largely influenced by genotype, as demonstrated in this study. The stratified analysis revealed that certain genetic defects, such as GUCY2C, SLC26A3, XIAP, BTK, TYMP, G6PC3, and CYBB deficiencies, were commonly identified in patients with LO-mIBD and frequently result in CD-like phenotypes. Additionally, monogenic disorders such as CEAS and familial/congenital diarrhea were prominent in patients with LO-mIBD. CEAS, typically observed in adolescents and adults with SLCO2A1 deficiency, mimics CD with severe and multiple small intestinal ulcers [29, 30]. Patients with congenital diarrhea (SLC26A3 or SLC9A3 mutations) and familial diarrhea (GUCY2C mutations) typically experience severe diarrhea and electrolyte imbalances early in life, with intestinal inflammation and obstruction often developing in late childhood or adulthood [31, 32]. These monogenic etiologies should be prioritized in patients with late-onset and refractory CD-like phenotypes, and adult gastroenterologists should fully understand the systemic manifestations and optimize clinical management.
Although most LO-mIBD cases exhibit features overlapping with CD, some distinct endoscopic, pathological, and imaging characteristics have been noted in certain patients with mIBD, which may aid in distinguishing them from those with classic IBD [20, 33, 34]. In addition, it is also important to recognize that patients with mIBD present with various GI phenotypes, even in late-onset populations. Examples include iBD-like isolated and round ulcers in those with haploinsufficiency of A20 (HA20) [35, 36] and celiac-like villous atrophy and lymphocyte infiltration in those with X-linked immune dysregulation, polyendocrinopathy, enteropathy syndrome (IPEX) or IPEX-like syndromes [37, 38]. Therefore, the GI phenotypes in late-onset mIBD should be comprehensively evaluated by integrating the details of endoscopic findings, histopathology, other laboratory tests, and systemic manifestations. Future research should be dedicated to clarifying the GI phenotypic spectrum to help optimize disease classification and enhance understanding of LO-mIBD. In addition, given the complex and genotype-driven systemic manifestations of mIBD, a multidisciplinary strategy is recommended for adult gastroenterologists in the screening and management of late-onset mIBD.
The clinical management of LO-mIBD remains complex, as patients with LO-mIBD also exhibit refractoriness to traditional treatment, which varies by genotype. In addition, those with CYBB defects present with milder phenotypes and better GI responses in LO-mIBD than in IO-mIBD, consistent with prior reports on CYBB-associated CGD colitis [38]. However, these differences may be influenced by factors such as drug dose, administration mode, and follow-up duration. Higher surgical resection rates in patients with LO-mIBD may reflect delayed diagnosis, more insidious disease progression, or reduced responsiveness to medications. For example, patients with SLCO2A1 or TYMP defects respond poorly to conventional therapy [39, 40], while those with GUCY2C defects may experience cumulative intestinal damage over time [31]. Variations in follow-up durations and in treatment strategies between pediatric and adult patients may further impact the outcomes, as surgical rates may increase over follow-up time and surgery is generally less recommended in children than in adults [41]. Nonetheless, due to the limited sample size—particularly for those undergoing HSCT—these findings regarding treatment and effectiveness remain exploratory and warrant validation in larger prospective cohorts.
In mIBD, the intestinal disease typically arises from immune and epithelial dysfunctions that vary by underlying genetic defects and shape both the clinical phenotype and treatment response [6, 42, 43]. In this study, LO-mIBD was more frequently associated with phagocyte-related immunodeficiencies and non-immune epithelial defects, while IO-mIBD was mostly linked to immune dysregulation pathways. This is consistent with a previous study that highlighted immunodeficiencies in earlier-onset mIBD populations [44]. Correspondingly, patients with LO-mIBD had significantly fewer recurrent infections and fevers than those with IO-mIBD, further supporting distinct immune pathogenic mechanisms between the two groups. Age-related variation in immune development may also play a role. These findings indicate that mIBD should be considered even in late-onset cases with less pronounced signs of immunodeficiency or infection susceptibility, despite prior studies emphasizing recurrent infections in very early-onset mIBD populations [45, 46]. Notably, LO-mIBD patients with phagocyte- or epithelial-related defects respond poorly to conventional therapies and lack effective alternatives [42, 47]. Targeting these pathways may hold therapeutic potential. Further functional studies are needed to confirm this hypothesis and support the development of targeted treatments.
This study focuses on late-onset mIBD for the first time. However, as the understanding of mIBD evolves, the definition and research of late-onset mIBD require ongoing refinement. In this study, we comparatively analyzed the characteristics of LO-mIBD and IO-mIBD cases. However, the age of onset, as well as the clinical and genetic features of mIBD, may follow a continuum. Patients with an onset between 2 and 16 years may exhibit intermediate characteristics. Future studies including a wider age range could help better delineate the disease spectrum and age-related trends in mIBD. Furthermore, the advancement and increased accessibility of genetic testing could largely facilitate the identification of mIBD, especially for cases with milder manifestations and later onset [48]. The interpretation of genetic variants in different studies may have been influenced by the rapid advancements in human genetics over time. Consequently, the definition of late-onset mIBD cases may evolve continuously, requiring ongoing data updates, enhanced comparability across studies, and further exploration of the correlation among specific variants, pathogenicity, and clinical outcomes.
This study had some limitations. First, the retrospective nature of the published data inherently introduced biases of missing data, but our preliminary sensitivity analyses indicated minor risks. Second, reliance on published case reports and series may lead to selection or publication bias. These reports tend to highlight rare and severe cases; therefore, the prevalence of LO-mIBD may be underestimated. Third, the heterogeneity among reports—disease understanding, diagnostic criteria and tools, and healthcare resources differed across eras and regions—potentially skews results toward regions or periods with advanced genetic testing and specialized expertise and introduces a risk of misclassification of GI phenotypes. Fourth, reliance on binary or categorical outcomes may have constrained the ability to capture complex clinical processes and details. Due to heterogeneous data and a small sample size, the robustness of the stratified analysis was limited, and a multivariate model including all confounding factors was not possible yet, which warrants further investigation in future studies. Finally, due to the retrospective design, the direct applicability of these identified features in LO-mIBD screening is limited and requires validation in future cohort studies. Collectively, these limitations could be addressed by future large-scale, prospective, and standardized studies to validate and extend our findings.
In conclusion, mIBD is a clinical challenge across various age groups, including adult gastroenterology. Late-onset mIBD remains highly heterogeneous and difficult to manage. Although genomic sequencing advances have facilitated the identification and reporting of LO-mIBD cases, a deeper understanding of this disease subgroup is still needed to establish timely and effective clinical management strategies. Our study delineates the clinical spectrum and underlying pathogenic mechanisms of LO‑mIBD—particularly those associated with relatively common genetic defects—providing insights for clinical practice and further research. Future studies on LO-mIBD involving larger sample sizes, diverse age subgroups, prospective controlled cohorts, as well as functional investigations are essential to validate and extend these observations and to optimize patient management.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplementary Material 1
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Jezernik G, Micetic-Turk D, Potocnik U. Molecular genetic architecture of Monogenic pediatric IBD differs from complex pediatric and adult IBD. J Pers Med. 2020, 10:243.10.3390/jpm 10040243 PMC 771225433255894 · doi ↗ · pubmed ↗
- 2Satam H, Joshi K, Mangrolia U et al. Next-Generation Sequencing Technology: Current Trends and Advancements. Biology (Basel). 2023, 12:997.10.3390/biology 12070997 PMC 1037629237508427 · doi ↗ · pubmed ↗