Molecular and morphological data support the synonymy of Muricanthusradix Gmelin, 1791 and Muricanthusambiguus Reeve, 1845 (Gastropoda, Muricidae)
Francisco Morinha, James Ernest, Ana Archer-Taveira, Ana M. Rocha, Robert T. Iwamasa

TL;DR
This study uses DNA and shell data to show that two species of murex snails are actually the same.
Contribution
The study provides the first genetic evidence supporting the synonymy of Muricanthusambiguus and Muricanthusradix.
Findings
DNA barcoding and ddRAD-seq revealed only two genetic clusters among the studied snails.
Mitochondrial DNA introgression suggests M.ambiguus and M.radix are the same species.
M.nigritus is genetically distinct from M.radix/ambiguus.
Abstract
The Muricanthusradix/ambiguus/nigritus complex includes species with a great diversity of shell shapes and shared habitats in various regions, which has raised questions and doubts about the current taxonomic classification of these species. Muricanthusnigritus, M.radix, and M.ambiguus are three similar-looking black and white murex found commonly on the west coast of North and South America. The wide variety of morphological patterns within and between these species makes the classification of specimens difficult by visual observation. To this day, controversy persists over whether M.radix and M.ambiguus are one or two distinct species. Molecular genetic data have helped clarify the taxonomic classification of many mollusk species in recent decades, contributing to a more accurate understanding of biodiversity and ecosystems. In this study, DNA barcoding and double digest…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
 Figure 1
Figure 1 Figure 2
Figure 2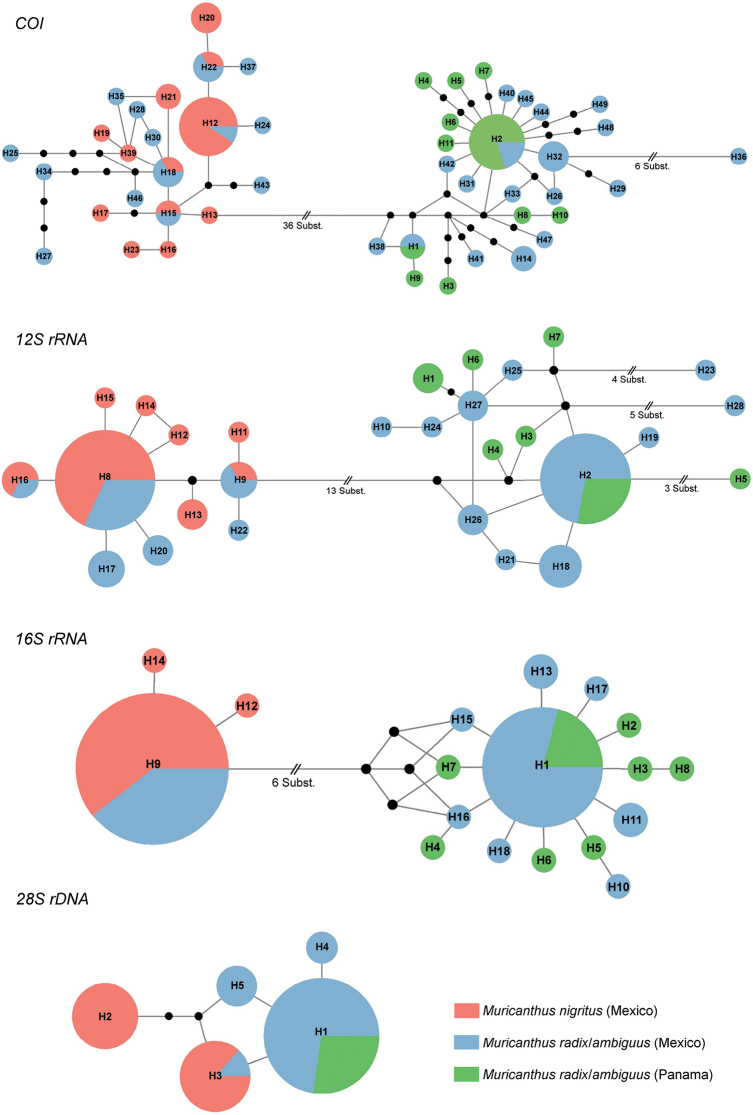 Figure 3
Figure 3 Figure 4
Figure 4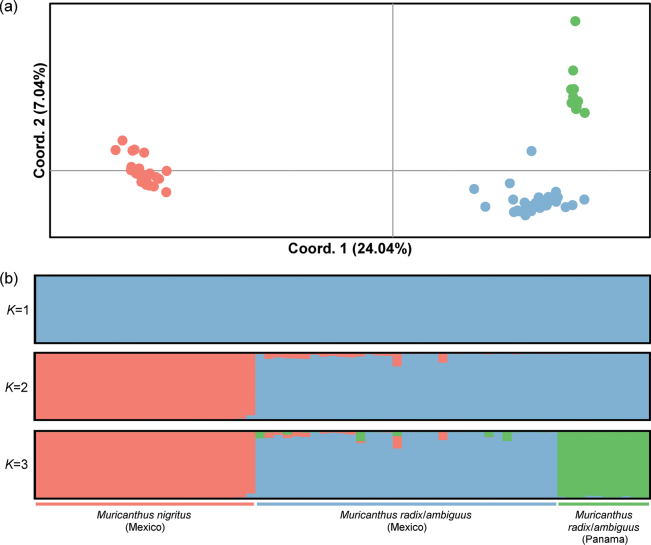 Figure 5
Figure 5| Gene region | Primer sequences (5′–3′) | Ta (°C) | Amplicon length (bp) | Reference |
|---|---|---|---|---|
|
| F: CWAATCAYAAAGATATTGGAAC | 50 | ~ 660 |
|
| R: AATATAWACTTCWGGGTGACC | ||||
|
| F: TGCCAGCAGYCGCGGTTA | 58 | ~ 560 | |
| R: AGAGYGRCGGGCGATGTGT | ||||
|
| F: CGCCTGTTTATCAAAAACAT | 60 | ~ 530 | Palumbi et al. 2001 |
| R: CCGGTCTGAACTCAGATCACGT | ||||
|
| F: TAGGTCGACCCGCTGAAYTTAAGCA | 58 | ~ 1430 | |
| R: AGCGCCATCCATTTTCAGG |
| Gene region | Best fit model | αa | Ib | Reference |
|---|---|---|---|---|
|
| HKY+G | 0.108 | – | Hasegawa et al. 1985 |
|
| HKY+I | – | 0.776 | Hasegawa et al. 1985 |
|
| HKY+I | – | 0.841 | Hasegawa et al. 1985 |
|
| HKY+I | – | 0.951 | Hasegawa et al. 1985 |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsMollusks and Parasites Studies · Aquatic Invertebrate Ecology and Behavior · Marine Biology and Ecology Research
Introduction
The Muricidae (Gastropoda, Muricoidea) is a family that includes nearly 1800 extant species distributed throughout the world, exhibiting a very high and complex taxonomic diversity (Barco et al. 2010; Russini et al. 2023). The genus Muricanthus Swainson, 1840 comprises five species proposed according to a morphology-based taxonomy (M.ambiguus Reeve, 1845; M.callidinus Berry, 1958; M.nigritus Philippi, 1845; M.radix Gmelin, 1791; and M.strausi Verrill, 1950) (Houart and Wiedrick 2021). The assignment of species names for different-looking, but similar, black and white murex has been the subject of confusion for many years due to considerable variations seen in the shell structure (Pittman 2023). The difficulty and confusion over the proper identification of the shells, whose binomial names were established as M.nigritus, M.radix, and M.ambiguus have led to the misidentification of many shells in scientific publications and in those discussed or sold on the internet (GBIF Secretariat 2023a, 2023b, 2023c; Iwamasa 2023; Pittman 2023).
Muricanthusnigritus is commonly found in the northern region of Baja California in the Gulf of California and as far west as San Carlos, Baja Sur, Mexico (Houart and Wiedrick 2021; Iwamasa 2023). This species is best known as a delicacy in the Asian market and has been extensively written about due to over harvesting (Cudney-Bueno and Rowell 2008). Muricanthusnigritus is characterized by a pyriform shape, a heavy shell; a high spire; a long siphonal canal; horn-shaped spines with many spiral cords and its four midsection spiral cords are small; a high number of varices, generally 6–12; and with a small or missing labral tooth (Houart and Wiedrick 2021; Iwamasa 2023).
Muricanthusradix is typically found from as far south as Peru to Panama and Costa Rica. Some specimens have been documented as far north as Manzanillo in the Sea of Cortez, on the west coast of central Mexico (Houart and Wiedrick 2021). Muricanthusradix is characterized by a globose, round, and heavy shell; short spire; short siphonal canal; numerous spiral cords with numerous open flowery spines; a high number of varices, generally 8–14; and has a labral tooth (Houart and Wiedrick 2021).
Muricanthusambiguus has been found along the southern west coast of Mexico, extending further south to Panama. Muricanthusambiguus is characterized by a pyriform-shaped shell; it weighs less than M.radix of the same size; has a higher spire; a longer siphonal canal; numerous spiral cords and numerous, larger, open, flowery spines (by comparison with the two former species); shell sometimes sharper, with fewer varices (by comparison with the two previous species), generally 6–10; and a labral tooth (Houart and Wiedrick 2021). Both M.radix and M.ambiguus are known to cohabitate in colonies (Houart and Wiedrick 2021; Pittman 2023). Considerable variations in shell structure occur within the species, making it difficult to identify based solely on morphology-based taxonomic descriptions (Pittman 2023). Since the early days of their descriptions to the present, there has been controversy over whether M.radix and M.ambiguus are one species or two (Pittman 2023).
Marine gastropods shell shape and size can be affected by genetic factors and several anthropogenic factors (e.g., chemical contamination and climatic changes) and non-anthropogenic factors (e.g., food availability, predatory pressure, salinity, temperature, hydrodynamics, desiccation, and substrate type) (e.g., Zimmerman and Pechenik 1991; Verhaegen et al. 2018; Harayashiki et al. 2020a, 2020b). The diversity and complexity of shell morphology of many gastropod species have made taxonomic classification difficult using morphometric data alone (e.g., Pfenninger et al. 2006; Kantor et al. 2017, 2022; Wu et al. 2022). This can lead to an overestimation or underestimation of the number of species, depending on local environmental conditions the regions from which the specimens are characterized (Whelan 2021). Correct species assignment is of paramount importance in various fields of biological research, including, but not limited to, biodiversity, evolution, conservation, and phylogeographic studies (De Carvalho et al. 2008). In this context, the application of molecular genetic approaches has proven to be very useful in complementing and/or overcoming the morphology-based taxonomy limitations in different organisms, including various mollusk species (Xu et al. 2024). DNA barcoding methodologies have been widely applied as a complement to traditional taxonomy in the classification of several gastropods (e.g., Borges et al. 2016; Ran et al. 2020). Nevertheless, the historical and/or recent hybridization events, along with the absence of a barcoding gap among closely-related species, may introduce bias in taxonomic classifications using DNA barcoding technology (Phillips et al. 2022). Advances in high-throughput Next Generation Sequencing (NGS) technologies have enabled the development of several cost-effective strategies for rapid genotyping of thousands of markers from the whole genome of any organism (da Fonseca et al. 2016). The application of these methodologies in biodiversity and conservation studies has increased significantly in recent years (Theissinger et al. 2023). Double digest restriction-site associated DNA sequencing (ddRAD-seq) is an NGS-based approach that allows for the discovery and genotyping of thousands of SNPs, even without the organism’s reference genome (Peterson et al. 2012). It is widely used in biological studies, including taxonomic classifications of a broad range of taxa (e.g., Peterson et al. 2012; Andrews et al. 2016; Ivanov et al. 2021; Paran et al. 2023). The development of integrative taxonomic approaches is crucial for enhancing and clarifying our understanding of species complexes (Pante et al. 2015; Karbstein et al. 2024).
In this study, we applied molecular techniques (DNA barcoding and ddRADseq) to clarify the discrepancies, confusion, and controversies associated with the taxonomic classification of three Muricanthus species. We obtained morphological and genetic data on M.nigritus, M.radix, and M.ambiguus specimens collected from different geographic areas for the first time. The implications of the new molecular findings for species assignment are discussed in light of the morphometric data of the specimens and classic taxonomic descriptions. The relationship between M.radix and M.ambiguus is clarified, highlighting the importance of developing future studies on species boundaries and population genetics.
Material and methods
Specimen collection
Samples included in this study were obtained opportunistically, through the collaboration with various fishing vessels between November 2021 and March 2023. A total of 80 Muricanthus specimens were sampled in different regions (Isla Cebaco, Panama; Jalisco, Mexico; and Magdalena Bay, Baja Sur, Mexico; Fig, 1) and collection sites (see approximate coordinates in Suppl. material 1). Photographs of six positions (dorsal, ventral, anterior, posterior, left and right sides) for each specimen were made using an iPhone 15 cell phone camera. Measurements on length, weight, and number of varices were recorded for all specimens. Weight measurements were made on cleaned shells, where encrustations were removed as best as possible using a sonic cleaner (VRN Ultrasonic Scaler, Model number VRN-A8) and dental tools. Cleaning time varied depending on the amount of encrustation, generally ranging from 25 minutes to 1 hour. The length measurements were made using a digital caliper from the apex to the tail end. Classification based on previous taxonomic descriptions (Houart and Wiedrick 2021) was carried out where possible. In addition, four samples of Hexaplexprinceps Broderip, 1833, and five samples of Phyllonotusregius Swainson, 1821, used as outgroups in the phylogenetic tree were collected in Panama (see approximate coordinates in Suppl. material 1). Approximately 500 mg of foot tissue was collected from the specimens, placed in glass vials containing absolute ethanol and stored at -20 °C in order to preserve the samples for molecular DNA analysis.
Geographic representation of sampling locations with some of the specimens collected at these sites (the dimensions are not to scale). Map was generated with ArcGIS 10.5 software (ESRI, Redland, USA, https://desktop.arcgis.com/en/).
DNA extraction, amplification, and Sanger sequencing
DNA was isolated from foot tissues using the Quick-DNA Miniprep Plus Kit (Zymo Research, Irvine, CA, USA) following the manufacturer’s protocols with some optimizations. The digestion mixture contained 180 µL of water, 180 µL of solid tissue buffer, 40 µL of proteinase K and 300 mg of tissue shredded with a scalpel. The incubation step was performed at 55 °C for 24 h. The DNA was eluted in 60 µL of elution buffer. DNA concentration and integrity were evaluated with the fluorimeter Qubit 3.0 using the Invitrogen Qubit dsDNA High-Sensitivity (HS) assay kit (Thermo Fisher Scientific, USA) and agarose gels of genomic DNA.
Three different regions of the mitochondrial DNA (cytochrome oxidase I – COI, 12S ribosomal RNA (rRNA) and 16S rRNA genes) and one fragment of the 28S rDNA nuclear gene were PCR-amplified with primers previously characterized (Table 1). Each reaction included 10 μL of 2× MyTaq HS Mix (Meridian Bioscience, USA), 2.5 µM of each primer, 20 ng of template DNA, and ultrapure DNase/RNase-free water to make a total volume of 20 μl. The PCR run consisted of 95 °C for 5 min followed by 40 cycles of 95 °C for 30 s, annealing at variable temperatures depending on the primers (Table 1) for 1 min, 72 °C for 30 s, and a final extension at 60 °C for 10 min. All samples were bi-directionally sequenced by Sanger sequencing. Sequences were visualized and edited using the Chromas software version 2.6.6 (https://technelysium.com.au/wp/chromas/). All sequences were submitted to GenBank (see accession numbers in Suppl. material 1).
Phylogenetic analyses
Median-joining networks were constructed for COI, 12S rRNA, 16S rRNA, and 28S rDNA haplotypes using the program NETWORK v. 5.0.1.1 (Bandelt et al. 1999). The program BEAST v. 2.6.0 (Bouckaert et al. 2014) was used to construct a Bayesian maximum clade credibility (MCC) tree using individual and concatenated sequences of the four gene regions characterized for the specimens analyzed. The software JMODELTEST v. 2.1.7 (Darriba et al. 2012) was applied to estimate the best model of sequence evolution for each gene region (Table 2). The proportion of invariable sites and the alpha shape parameter (α) used to construct the phylogenetic tree are present in Table 2. The strict clock and Yule model priors were applied, with a normal distribution for the rate prior, letting the program estimate the mutation rates. The software run consisted of 100 million steps, with a sampling of the chains every 1,000 steps and a burn-in of 10%. The proper convergence and mixing of the chains, and effective sample sizes (ESS) were evaluated with TRACER v. 1.7.1 (Rambaut et al. 2018). The consensus tree was visualized and edited with FIGTREE v. 1.4.4 (Rambaut 2018).
Species delimitation
The DNA-based species delimitation analyses were conducted using the ASAP (Assemble Species by Automatic Partitioning) distance-based method (Puillandre et al. 2021). The COI datasets were used for the ASAP analyses through its web server (https://bioinfo.mnhn.fr/abi/public/asap/, accessed on 22 March 2025), applying the default settings and the Kimura two-parameter (K2P) substitution model. The first analysis included the complete COI dataset with the 89 sequences including the outgroup species (M.nigritus, M.radix, M.ambiguus, H.princeps, and P.regius). The second analysis included only the COI sequences obtained from the 80 Muricanthus specimens.
ddRADseq library preparation, sequencing, and data analysis
The ddRADseq analysis included only those specimens from which it was possible to obtain sufficient amounts of DNA and that passed the sample quality controls recommended for the protocol. Thus, a representative sample of all specimens and species were included (n = 61). Six random samples were analyzed in duplicate and used as quality and reproducibility controls for the protocol. The ddRAD libraries were prepared following a previously published protocol (Daguin-Thiébaut et al. 2021). The libraries were sequenced in 75-bp paired-end reads using the Illumina NextSeq 550 Sequencing System. Raw reads were processed using the software pipeline STACKS v. 1.44 (Catchen et al. 2011) for demultiplexing, quality filtering, de novo locus assembly, and SNP screening. The process_radtags module was used to remove barcodes and to discard low-quality reads (Catchen et al. 2011). The ustacks module was used to merge filtered RADtags into loci within each specimen after an evaluation and selection of the best combination of parameters (-m 2, -M 2, -n 2), following the recommendations and guidelines of previous studies (Paris et al. 2017; Rochette and Catchen 2017). Only loci genotyped in at least 80% of individuals (−r 0.8) were considered. The SNPs obtained after de novo locus assembly were filtered for some parameters with TASSEL v. 5. Namely, loci with heterozygosity > 0.8 and a minor allele frequency (MAF) < 5% were excluded.
A total of 3692 individual multilocus genotypes obtained from ddRADseq were used to group specimens according to their SNP profiles. Specimens were clustered according to the total genetic variation using a Principal Coordinate Analysis (PCoA) conducted in GENEALEX v. 6.5 (Peakall and Smouse 2012). The most likely number of genetic clusters using a Bayesian clustering approach was obtained using the program STRUCTURE v. 2.1 (Pritchard et al. 2000). The model of correlated allele frequencies and admixture ancestry without sampling localities as priors was used. The software run consisted of ten replicates for each K value from 1 to 4 with 1,000,000 Markov chain Monte Carlo (MCMC) generations after a burn-in of 100 000 iterations. The results of the assignment probabilities of individuals (q) to each cluster were obtained through the CLUMPAK server (Kopelman et al. 2015), considering an MCL threshold for similarity scores of 0.90. The optimal K value was inferred with STRUCTURE HARVESTER using the Evanno method (Evanno et al. 2005; Earl and von Holdt 2012). We assessed pairwise genetic differentiation between the clusters identified using the F_ST_ estimator of Weir and Cockerham (1984). The F_ST_ values were estimated using the analysis of molecular variance (AMOVA) with 9999 permutations, as implemented in GENALEX v. 6.5.
Results
Morphological data
The 80 Muricanthus specimens were classified based on photographs and morphological descriptions published by Houart and Wiedrick (2021). Twenty-five individuals were assigned to M.nigritus, 14 were identified as M.radix, 32 were classified as M.ambiguus, and nine were identified as M.radix/ambiguus (Suppl. material 1). These nine specimens had morphological characteristics common to M.radix and M.ambiguus, making it impossible to assign them to a specific species. Examples of the different species and specimens where correct taxonomic identification is difficult are illustrated in Fig. 2. The morphometric data and identification outcomes of all collected Muricanthus specimens are reported in Suppl. material 1. Photographs of six representative examples of specimens for each of the three geographical areas are shown in Suppl. material 2: figs S1, S2 (Magdalena Bay, Baja Sur, Mexico), Suppl. material 2: fig. S3 (Jalisco, Mexico) and Suppl. material 2: fig. S4 (Isla Cebaco, Panama).
Dorsal and ventral views of shells of M.nigritus (1N and 23N), M.radix (2A, 6A, V5, C1 and C4), M.ambiguus (5A, 12A, MR3, V6, C5, C6, C8, C12) and individuals that are difficult to classify M.radix/ambiguus (V4 and V11). The measurements given are shell lengths. More specimens and different views of the shells are presented in Suppl. material 2: figs S1–S4.
Haplotype networks and phylogenetic inferences
The partial sequences of mtDNA COI (662 bp), 12S rRNA (approx. 560 bp), 16S rRNA (532 bp) and nuclear 28S rDNA (approx. 1430 bp) were successfully sequenced for the 80 Muricanthus specimens analyzed. As expected, a high number of haplotypes were identified in the mitochondrial DNA sequences (49 for COI, 28 for 12S rRNA, and 18 for 16S rRNA) and a smaller number in the 28S rDNA nuclear sequences (5 haplotypes). The four median-joining statistical parsimony networks revealed that various mtDNA and nuclear haplotypes are shared by specimens of M.nigritus and M.radix/ambiguus collected in Mexico (Fig. 3). Several specimens of M.radix/ambiguus from Mexico exhibited specific haplotypes of M.nigritus; however, none of the M.nigritus specimens analyzed contained any of the M.radix/ambiguus specific haplotypes identified, considering the species identification determined by shell morphology (Fig. 3; Suppl. material 1). No shared haplotype was detected between M.nigritus from Mexico and M.radix/ambiguus from Panama (Fig. 3). Specimens of M.radix/ambiguus from Mexico and Panama also share several haplotypes (Fig. 3). Two major haplogroups are clearly separated in the COI, 12S rRNA, and 16S rRNA networks, one corresponding to M.nigritus and M.radix/ambiguus from Mexico, and the other corresponding M.radix/ambiguus from Mexico and Panama. The Bayesian maximum clade credibility tree obtained using concatenated sequences confirmed the existence of two main, well-supported clades: one including M.nigritus and M.radix/ambiguus haplotypes from Mexico, and the other corresponding to M.radix/sambiguus haplotypes from Mexico and Panama (Fig. 4). The individual trees of the three mtDNA regions analysed show very similar results (Suppl. material 2: figs S5–S7). The 28S rDNA tree also shows some differentiation, but less pronounced (Suppl. material 2: fig. S8).
Median-joining networks of all haplotypes identified for the four gene regions studied. Haplotypes are represented by different circles with a size proportional to their frequency (see Suppl. material 1). All cases in which more than one nucleotide substitution occurs between haplotypes are indicated, and all other haplotypes are separated by a single nucleotide change. Solid black circles represent unsampled haplotypes.
Bayesian maximum clade credibility tree of the concatenated sequences (COI, 12S rRNA, 16S rRNA, 28S rDNA). Posterior probabilities for the nodes of the most divergent clades are presented.
Species delimitation
The best species partition based on the ASAP-score split our complete COI dataset (including the outgroup species) in 4 hypothetical species (threshold distance = 3.88%, Suppl. material 2: fig. S9a). This analysis split the samples of the three Muricanthus species into only two hypothetical species. The ASAP analysis of the COI dataset of Muricanthus samples alone also suggests the presence of only two hypothetical species, according to the best ASAP-score (threshold distance = 3.34%; Suppl. material 2: fig. S9b).
Estimation of genetic clusters from ddRADseq dataset
The ddRADseq approach was applied to confirm the existence of only two phylogenetic groups among M.nigritus, M.radix, and M.ambiguus through a more in-depth molecular analysis. Our de novo assembly with ustacks showed that the number of ddRAD tags recovered from the samples analyzed varied from 119321 to 213181 with a mean coverage of 4×. These ddRAD-tags contained 996260 SNPs. After SNP filtering, the ddRASseq protocol allowed the selection of 3692 SNPs used to cluster the specimens according to their genetic divergence/differentiation (Suppl. material 3). The PCoA and Bayesian clustering approaches showed very similar results (Fig. 5a, b). PCoA separated the M.nigritus, M.radix/ambiguus (Mexico) and M.radix/ambiguus (Panama) (Fig. 5a). The optimal number of clusters inferred in the Bayesian clustering analysis without sample location prior was K = 2, corresponding to M.nigritus and M.radix/ambiguus (Mexico and Panama) (Fig. 5b). However, the simulations for K = 3 also made it possible to differentiate M.radix/ambiguus (Mexico) and M.radix/ambiguus (Panama) (Fig. 5b). It was not possible to differentiate any further groups within these three final clusters. The pairwise genetic differentiation between cluster 1 (M.nigritus) and cluster 2 (M.radix/ambiguus, Mexico) was F_ST_ = 0.193, p < 0.001, between cluster 1 and cluster 3 (M.radix/ambiguus, Panama) was F_ST_ = 0.242, p < 0.001, and between cluster 2 and cluster 3 was F_ST_ = 0.107, p < 0.001. The multilocus genotype data clearly differentiated the M.nigritus specimens from the M.radix/ambiguus specimens. None of the molecular techniques applied in this work made it possible to differentiate specimens identified as M.radix and M.ambiguus based on morphology.
Characterization of genetic clusters obtained using SNP data a PCoA plot of genetic distances estimated using the method of Smouse and Peakall (1999)b Clusters obtained from the Bayesian clustering analysis without prior information on sample location. Each vertical line corresponds to one individual. The cluster assignments obtained for each K tested are represented with different colors, where the optimal number of clusters was estimated for K = 2.
Nomenclatural act
Muricanthusambiguus (A.L. Reeve, 1845) is here synonymized with Muricanthusradix (Gmelin, 1791) considering the lack of genetic differentiation between the morphologically assigned specimens.
Discussion
The genus Muricanthus includes some species that have undergone several taxonomic reclassifications over the last few centuries (Houart and Wiedrick 2021). The difficulties in the proper taxonomic classification of M.radix and M.ambiguus have been recognized for many years, most recently by Pittman (2023), who pointed out that it is not possible to accurately assign specimens due to the considerable morphological variation seen in their shell structure. Their study hypothesizes that M.ambiguus may be a transitional form of M.radix from the south and M.nigritus from the north (Pittman 2023). The possibility that M.nigritus and M.ambiguus are northern subspecies of M.radix is also highlighted in some publications (Fischer et al. 1995; Escamilla-Montes et al. 2018). Muricanthusnigritus is the easiest to identify taxonomically due to its horn-shaped spines, with the spines on the mid-section of the last whorl being shorter than those on other sections and absent in M.radix or M.ambiguus. Muricanthusnigritus also has a very short or missing labral tooth (Houart and Wiedrick 2021; Iwamasa 2023). In the case of specimens of M.radix and M.ambiguus, the spines are nearly identical and flowery, and both also have a noticeable labral tooth (Houart and Wiedrick 2021). There are differences in spire height, siphonal canal length, and the number of varices, but the variations in morphological structure among the specimens make it difficult to assign some to either species. While there are distinct examples of specimens that can be classified taxonomically as either M.radix or M.ambiguus, a larger sample of the species reveals difficulties in their assignment, as shown by the numerous examples reported in this study. These species are also commonly misclassified in online biodiversity databases (GBIF Secretariat 2023a, b, c). The high diversity of shell shapes within various mollusk taxa has raised several questions about the possible overestimation or underestimation of the number of species in the classifications based solely on shell morphometrics (Shea et al. 2011). It is well known that environmental and genetic factors both affect shell structure (Gemmell et al. 2018; Clark et al. 2020; Iwamasa 2023). The Muricanthus specimens collected from the three different geographical areas showed differences in size, shape, number of varices, and condition, likely due to the environmental conditions in which they developed. From the earliest stages of veliger formation to juvenile and adult stages, the shell continues to grow volumetrically and can produce new varices (Cudney-Bueno et al. 2008; Iwamasa 2023). Environmental conditions can affect the morphology of the shell, resulting in similar-looking specimens between different species and regions. In addition, we observed that many adult shells also show signs of erosion due to predation and aging, further complicating identification. Given the subjectivity and difficulties in the species identification of several Muricanthus specimens, an integrative approach with the inclusion of several molecular markers can be an effective strategy to address these issues (Nocella et al. 2024).
The analysis of variations in mitochondrial and nuclear DNA has helped to clarify various taxonomic ambiguities in a wide diversity of mollusk taxa (Xu et al. 2024). In our study, the analysis of three mitochondrial sequences (COI, 12S rRNA, and 16S rRNA) clearly differentiated the specimens into two groups. Interestingly, one of the groups included individuals of M.nigritus (Mexico) and M.radix/ambiguus (Mexico) and another included individuals of M.ambiguus and M.radix (Mexico and Panama). The analysis of the nuclear 28S rDNA sequences revealed similar results, although the differentiation was lower. This is expected, as the mutation rates of nuclear DNA sequences are lower than those of mitochondrial DNA (Duda 2021). The fact that several haplotypes are shared between M.nigritus and M.radix/ambiguus may indicate the possible occurrence of historical and/or recent hybridization between these species. Our data suggest a potential asymmetrical hybridization process in the sympatric areas, since only the M.ambiguus/radix species presented M.nigritus haplotypes. Mitochondrial DNA introgression has been reported in several organisms, including mollusk species (e.g., Toews and Brelsford 2012; Yu et al. 2014; Garcia-Souto et al. 2024). Introgressive hybridization is an evolutionary process involving different biological processes, such as adaptation and response to environmental changes, speciation, and regulation of biodiversity levels (Dowling and Secor 1997).
None of the “classical” mitochondrial and nuclear DNA markers differentiated M.radix from M.ambiguus, highlighting the presence of several common haplotypes between the specimens from Mexico and Panama. This does not support the classification of M.radix and M.ambiguus as two distinct species based on morphometric data, although the sharing of mtDNA haplotypes can happen in some cases when very recent evolutionary processes of species divergence are involved, or where ancestral mtDNA polymorphisms are retained in recently diverged clades (Moritz et al. 1987; Galtier et al. 2009). The DNA-based species delimitation analyses support the data obtained from the Bayesian phylogenetic inferences, highlighting that the Muricanthus samples are split into only two species.
In this context, double digest restriction-site associated DNA sequencing (ddRAD-seq) was used for a more in-depth analysis of the genetic differentiation among the Muricanthus individuals. Several studies on species classification have already shown that this methodology is an efficient approach to clarify doubts left by “classic” DNA markers (e.g., Glon et al. 2021; Kim and Roe 2021; Obiol et al. 2023). The principal component and Bayesian clustering analysis of the new SNPs discovered in this study showed very similar results, allowing the specimens to be grouped into three clusters. Specimens of M.nigritus were clearly differentiated from specimens of M.radix/ambiguus. It was also possible to differentiate specimens of M.radix/ambiguus (Mexico) from specimens of M.radix/ambiguus (Panama) in both analyses. Once again, it was not possible to differentiate any cluster/individual between M.radix and M.ambiguus. Furthermore, Bayesian analysis suggests that the optimal number of clusters is K = 2, where M.radix (Mexico and Panama) and M.ambiguus (Mexico and Panama) are part of a single cluster. This finding indicates that the genetic differentiation observed between M.radix/M.ambiguus from Panama and Mexico may be associated with the geographical distance between these populations. Limitations in the dispersal potential between distant geographical areas and/or discontinuity of suitable habitats can explain the genetic differentiation observed. Although the analysis of the genetic data did not allow us to differentiate between M.radix and M.ambiguus in Mexico or in Panama, we noticed that the shells of the Panama specimens are generally smaller, at least in this collection site. These variations in shell size may be caused by genetic or environmental factors. Variations in shell size in the same gastropod species between different regions have been reported in several studies (e.g., Irie 2006; Cudney-Bueno et al. 2008; Malve et al. 2018; Matos et al. 2020; Iwamasa 2023). Within this context, our study also raises the question of whether M.callidinus Berry, 1958 and M.strausi Verrill, 1950 are truly distinct and different Muricanthus species. The inclusion of molecular data for the proper classification of these species can be an effective method of resolving taxonomic issues and clarifying their phylogenetic positions among the Muricanthus species complex.
Conclusions
This is the first study to integrate molecular techniques into the process of identifying Muricanthus specimens. Our molecular analysis reveals that M.radix and M.ambiguus are not genetically distinct, despite being classified as separate species based on morphology. In addition, a possible occurrence of mtDNA introgression was observed between M.nigritus and M.radix/ambiguus, suggesting possible hybridization processes between these species. The morphology of all the Muricanthus specimens studied from the three largely separated areas showed the problems of species identification based on the morphometric descriptions derived by the original authors and many others since then. The molecular approaches applied in this study highlighted that a morphology-based classification alone may be erroneous in this species complex. The data presented in this research indicate that M.ambiguus is a synonym of M.radix. Although it is possible to differentiate the M.nigritus specimens studied at the morphological and genetic level from M.radix/ambiguus, further research of more specimens from different regions is needed to clarify the possible hybridization between M.nigritus and M.radix/ambiguus, and the species boundaries. The high level of phenotypic plasticity in Muricanthus species is evidenced in this work and in previous studies (Iwamasa 2023; Pittman 2023), which highlights the importance of using an integrative taxonomy approach in future studies on this subject. Our study indicates the need to develop further genetic studies not only to clarify phylogenetic relationships between other Muricanthus species, but also to understand their diversity, genetic structure, evolutionary processes, and species distribution. This is crucial for implementing targeted conservation strategies for these species and their habitats, as they are exposed to several anthropogenic pressures.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Andrews KR Good JM Miller MR Luikart G Hohenlohe PA (2016) Harnessing the power of RA Dseq for ecological and evolutionary genomics. Nature Reviews.Genetics 17(2): 81–92. 10.1038/nrg.2015.28PMC 482302126729255 · doi ↗ · pubmed ↗
- 2Bandelt HJ Forster PRöhl A (1999) Median-joining networks for inferring intraspecific phylogenies.Molecular Biology and Evolution 16(1): 37–48. 10.1093/oxfordjournals.molbev.a 02603610331250 · doi ↗ · pubmed ↗
- 3Bandyopadhyay PK Stevenson BJ Ownby JP Cady MT Watkins M Olivera BM (2008) The mitochondrial genome of Conustextile, cox I–cox II intergenic sequences and Conoidean evolution.Molecular Phylogenetics and Evolution 46(1): 215–223. 10.1016/j.ympev.2007.08.00217936021 PMC 2718723 · doi ↗ · pubmed ↗
- 4Barco A Claremont M Reid DG Houart R Bouchet P Williams ST Cruaud C Couloux A Oliverio M (2010) A molecular phylogenetic framework for the Muricidae, a diverse family of carnivorous gastropods.Molecular Phylogenetics and Evolution 56(3): 1025–1039. 10.1016/j.ympev.2010.03.00820226866 · doi ↗ · pubmed ↗
- 5Borges LM Hollatz C Lobo J Cunha AM Vilela AP Calado G Coelho R Costa AC Ferreira MSG Costa MH Costa FO (2016) With a little help from DNA barcoding: Investigating the diversity of Gastropoda from the Portuguese coast.Scientific Reports 6(1): 20226. 10.1038/srep 2022626876495 PMC 4753432 · doi ↗ · pubmed ↗
- 6Bouckaert R Heled JKühnert D Vaughan T Wu CH Xie D Suchard MA Rambaut A Drummond AJ (2014) BEAST 2: A software platform for Bayesian evolutionary analysis. P Lo S Computational Biology 10(4): e 1003537. 10.1371/journal.pcbi.1003537 PMC 398517124722319 · doi ↗ · pubmed ↗
- 7Catchen JM Amores A Hohenlohe P Cresko W Postlethwait JH (2011) Stacks: building and genotyping loci de novo from short-read sequences. G 3: Genes|Genomes|Genetics 1(3): 171–182. 10.1534/g 3.111.000240 PMC 327613622384329 · doi ↗ · pubmed ↗
- 8Clark MS Peck LS Arivalagan J Backeljau T Berland S Cardoso JCR Caurcel C Chapelle G De Noia M Dupont S Gharbi K Hoffman JI Last KS Marie A Melzner F Michalek K Morris J Power DM Ramesh K Sanders T SillanpääK Sleight VA Stewart-Sinclair PJ Sundell K Telesca L Vendrami DLJ Ventura A Wilding TA Yarra T Harper EM (2020) Deciphering mollusc shell production: The roles of genetic mechanisms through to ecology, aquaculture and biomimetics.Biological Reviews of the Cambridge Philosophical Society 95(6): 1812–1837. 10.1111/brv.1264032737956 · doi ↗ · pubmed ↗