The Role of Metabolic Testing in the Diagnostic Evaluation of Adult NORSE: A Retrospective, Single‐Centre Study
Jennifer Kilmer, George Ransley, Elaine Murphy, Michael G. Hanna, Robert D. S. Pitceathly, Sanjeev Rajakulendran, Chiara Pizzamiglio

TL;DR
This study examines the usefulness of metabolic testing in diagnosing the cause of NORSE in adults and finds limited utility in identifying inherited metabolic disorders.
Contribution
The study provides insights into distinguishing mitochondrial disease-related RSE from cryptogenic NORSE using clinical features.
Findings
Extensive metabolic testing did not identify inherited metabolic disorders as causes of NORSE.
Three patients with RSE had primary mitochondrial disease, not meeting NORSE criteria.
Criteria to differentiate PMD-related RSE from cNORSE include multisystem features and family history.
Abstract
New‐onset refractory status epilepticus (NORSE) is a diagnostically challenging and severe epileptic presentation in which aetiology is an important predictor of outcome. This retrospective study aimed to investigate the utility of metabolic screening to determine the underlying cause in 42 patients with suspected NORSE, admitted to The National Hospital for Neurology and Neurosurgery, London, between 2004 and 2021. Demographic, clinical, biochemical, and molecular data were collected. Sixty‐two per cent of the cohort was classified as cryptogenic (cNORSE), while 38% had symptomatic NORSE (sNORSE). Despite extensive investigations (100 metabolic‐related tests were performed among the 42 cases), inherited disorders of metabolism were not identified as causes for NORSE. Nevertheless, three patients with refractory status epilepticus (RSE), who did not fulfill the diagnostic criteria for…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
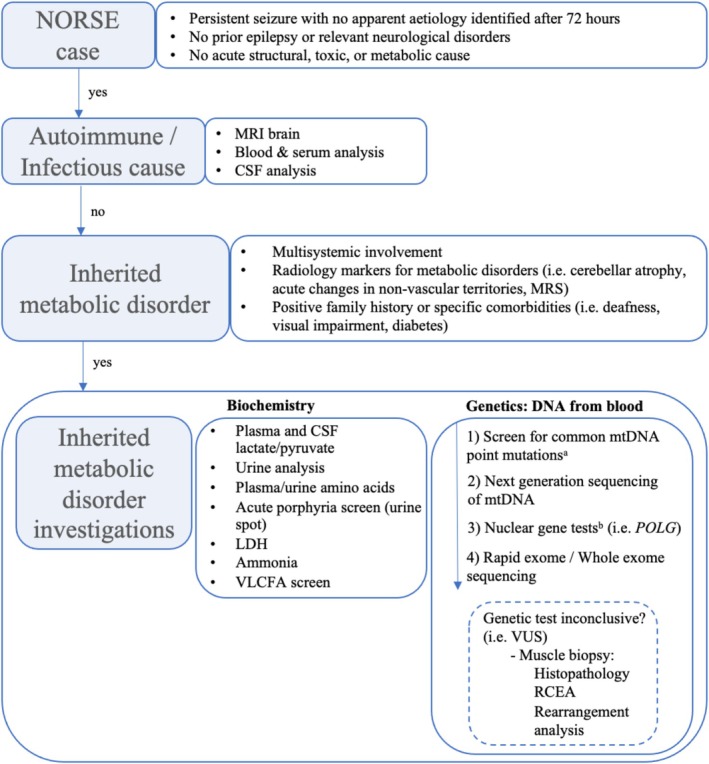 FIGURE 1
FIGURE 1| Test name | Completed | Abnormal |
|---|---|---|
|
|
| |
|
| ||
| Aminoacidopathies/Organic acidemias/Urea cycle defects ( | ||
| Urine/Plasma amino acid profile | 19 | 1 (5.3) |
| Urine organic acids | 9 | 3 (33.3) |
| Acyl carnitine profile | 6 | 0 (0) |
| Methylmalonic acid | 2 | 0 (0) |
| Disorders of energy metabolism ( | ||
| Mitochondrial genetic testing | ||
| mtDNA common point mutations | 10 | 0 (0) |
| Maintenance panel | 5 | 0 (0) |
| Whole mtDNA sequencing | 2 | 0 (0) |
| Whole exome sequencing (WES) | 2 | 0 (0) |
| Muscle biopsy | 6 | 1 (16.7) |
| Urine/plasma creatine and guanidinoacetate | 4 | 0 (0) |
| Respiratory chain enzyme analysis (RCEA) in muscle | 3 | 0 (0) |
| FGF21 | 3 | 0 (0) |
| Cofactor‐related disorders ( | ||
| CSF pterins | 5 | 0 (0) |
| CSF serotonergic/dopaminergic metabolites | 4 | 0 (0) |
| Biotinidase | 3 | 0 (0) |
| ɑ‐AASA | 1 | 0 (0) |
|
| 1 | 0 (0) |
| Purine/Pyrimidine disorders ( | ||
| Urine purines/pyrimidines | 4 | 0 (0) |
| Lesch–Nyhan genetic testing ( | 1 | 0 (0) |
| Peroxisomal/lysosomal disorders ( | ||
| White cell enzyme activity | 5 | 0 (0) |
| Very long chain fatty acid profile | 4 | 0 (0) |
| Bile acid intermediates | 1 | 0 (0) |
|
| ||
| Ammonia | 25 | 16 (64) |
| Glucose | 24 | 6 (25) |
| CSF lactate | 20 | 2 (10) |
| Serum lactate | 19 | 3 (15.8) |
| Serum folate | 15 | 4 (26.7) |
| Homocysteine | 10 | 3 (30) |
- —Medical Research Council 10.13039/501100000265
- —Lily Foundation 10.13039/501100022186
- —Muscular Dystrophy UK 10.13039/100011724
- —Rosetrees Trust 10.13039/501100000833
- —LifeArc 10.13039/100012357
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsEpilepsy research and treatment · Ion channel regulation and function · Mitochondrial Function and Pathology
Introduction
1
New‐onset refractory status epilepticus (NORSE) is defined as prolonged seizure activity refractory to first and second‐line antiseizure medications (ASMs), with no obvious aetiology determined in the first 72 h, without a prior history of epilepsy [1]. NORSE has a high mortality rate (16%–27%), and poor functional outcomes in up to two‐thirds of survivors [2].
A key determinant of outcome in status epilepticus is the underlying cause, which underscores one of the principal challenges of managing NORSE, where the cause remains unknown in up to 50% [3]. Such ‘cryptogenic’ NORSE (cNORSE) cases potentially run a more severe course [2].
While inherited metabolic disorders (IMDs), particularly primary mitochondrial diseases (PMDs), are considered in the diagnostic work‐up of NORSE typically when autoimmune and infectious causes are excluded, their role in adult‐onset NORSE is unclear and their prevalence has not been systematically studied [4, 5]. This often entails extensive genetic and biochemical tests in blood, CSF, and muscle.
This study aims to address this gap by evaluating the utility of metabolic screening in a cohort of adult‐onset NORSE to enhance the diagnostic process and inform clinical decision making.
Methods
2
This retrospective study reviewed all cases aged 16 years or older admitted to The National Hospital for Neurology and Neurosurgery (NHNN) neurological intensive care unit (ICU) with refractory SE (RSE) between January/2004 and December/2021. Of these, we have subsequently identified cases with NORSE as defined by Hirsch et al. [1] Cases were further categorised into symptomatic NORSE (sNORSE) or cNORSE based on the presence or absence of a final diagnosis. Twenty‐five NORSE cases were previously reported elsewhere [6].
Patient data, including demographics, clinical variables, outcomes and test results, were extracted from hospital records. Outcomes at discharge and 1‐year follow‐up were rated using the modified Rankin Scale (mRS) for neurological disability [7]. Mortality rates were ascertained at three timepoints: during admission, at 1‐year follow‐up, and through to July/2022 (end of observation). Results of a comprehensive battery of second‐line IMD investigations were designated normal or abnormal based on standard reference ranges available from the NHNN laboratory. The clinical relevance of these tests was established based on clinician opinions documented in the final report. The decision to conduct second‐line IMD investigations, including the type of investigations, was made by the clinician on a case‐by‐case basis.
Results throughout this study are presented as median (interquartile range [IQR]) or mean ± standard deviation (SD). Data were compared using Mann Whitney U test or Pearson's chi‐squared test. A p value < 0.05 was considered significant. Analyses were performed with SPSS (v. 19.0 for Windows; SPSS Inc.).
This study was submitted to the Clinical Audit and Quality Improvement Subcommittee at University College London Hospitals Trust. Informed consent was not required as the data were collected as part of routine clinical practice.
Results
3
Demographics and Clinical Characteristics
3.1
Demographics and clinical characteristics are summarised in Tables S1–S4, Figure S2. A total of 89 patients with SE were admitted to the NHNN ICU between January/2004 and December/2021. Of these, 73 were classified as RSE and 42 (57.5%) met the criteria for NORSE (Figure S1). An aetiology was determined in 16 cases (38%), while the remaining 26 cases (62%) were classified as cNORSE.
In the NORSE cohort there were more females (59.5%) and the median age was 28 years. The median ICU stay was 72 days, and the hospital stay was 105 days. Outcomes were generally poor, with 83.4% of patients having an mRS score ≥ 4 at discharge, indicating moderately severe disability or worse. The in‐hospital mortality rate was 15.4%, which increased to 20.5% at 1 year. There were no significant differences between cNORSE and sNORSE cases in terms of mortality rates and mRS outcomes.
Metabolic Investigations
3.2
IMD investigations were performed in 50% of patients (n = 21). Of these, PMD was the most common disorder (10/21). These tests were more commonly performed in cNORSE compared to sNORSE (p < 0.001 for IMD, and p = 0.04 for PMD), Figure S4, Table S5.
Table 1 outlines the type and frequency of metabolic investigations completed in our NORSE cohort, grouped as defined by Almannai et al. [8] Most metabolic test results were normal, and all cases of abnormal results were deemed clinically irrelevant. Despite constituting a considerable proportion of the average work‐up (100 IMD second‐line investigations), none of these tests contributed to identifying the NORSE aetiology in our cohort. Muscle histopathology and respiratory chain enzyme (RCE) analyses did not reveal any features of metabolic pathology, including mitochondrial dysfunction.
Of the 100 IMD investigations, 21 were genetic tests performed in 10 patients (24%). PMD testing constituted the vast majority, accounting for 19/21 genetic tests performed in 10 patients (nine with cNORSE and one with a final diagnosis of autoimmune sNORSE). All tests were negative. Tests included: common mtDNA pathogenic point mutations (m.3243A>G, m.8344A>G, and m.8993T>G/C) (n = 10), panel‐based testing of 23 relevant mitochondrial nuclear‐encoded genes including POLG (n = 5), whole sequencing of the mitochondrial genome (n = 2), and whole exome sequencing (n = 2). Metabolic, non‐mitochondrial genetics included analysis of HPRT (Lesch–Nyhan syndrome) (n = 1) and FOLR1 (cerebral folate transport deficiency) (n = 1).
Additional non‐metabolic genetic tests (mostly linked with epileptic encephalopathies) were performed in eight patients, all with negative results (Table S5).
Inherited Metabolic Disorders in RSE
3.3
While no IMD was identified in the NORSE cohort, three PMD cases (out of seven tested) were found in the RSE group (Figure S1), none meeting NORSE criteria. One case had the m.3243A>G, MT‐TL1 pathogenic variant in mtDNA (heteroplasmy: 17% in blood, 89% in urine). The phenotype was consistent with myoclonic epilepsy with ragged red fibres (MERRF)/Mitochondrial encephalomyopathy, lactic acidosis and stroke‐like episodes (MELAS) overlap. The second one had autosomal recessive pathogenic variants in POLG (c.1399G>A, p.Ala467Thr). A third patient showed multiple mtDNA deletions in muscle, suggesting a nuclear gene defect of mtDNA maintenance, but no genetic confirmation was detected. Interestingly, differences between the PMD‐related RSE and the 10 NORSE cases where PMD was ruled out through testing can be noted.
Firstly, the PMD‐related RSE had a previous history of epilepsy, indicating that the initial presentation of PMD with NORSE is uncommon. PMD imaging (Figure S3) revealed pronounced bilateral cerebellar atrophy at the SE onset‐MRI, thus suggesting an ongoing chronic process besides the acute SE (Figure S3b). Notably, metabolic infarcts were observed in two cases (m.3243A>G and POLG pathogenic variant) (Figure S3a,c). These were in contrast with the normal or nonspecific changes observed in the cNORSE cases (Figure S3e), or with the infectious‐related sNORSE (Figure S3d).
Similarly, the muscle biopsy of the POLG‐related PMD demonstrated abnormalities consistent with mitochondrial alterations, including ragged red fibres with reduced or absent COX activity, while none of the six biopsies in the cNORSE cases exhibited such features.
Discussion
4
This study evaluated 42 NORSE patients, the youngest aged 18, with 26 (62%) classified as cryptogenic. Investigations conducted after excluding infectious and autoimmune causes did not identify any metabolic diagnosis in our cohort.
IMD, a rare and diverse group of genetic disorders affecting metabolic pathways, is reported as a ‘rare’ cause of SE, and a list of IMD‐associated genes, mostly including mitochondrial genes, has been described [4]. Specific cases of NORSE involve mtDNA mutations (e.g., MT‐TF1) and nDNA mutations (e.g., POLG, DNM1L, FASTKD2), though most reports involve single cases, primarily in children [9, 10, 11, 12]. Among these IMDs, some potentially treatable causes of NORSE, such as CAD deficiency, should not be overlooked [13].
PMD‐related RSE cases differed from the cNORSE cohort who underwent mitochondrial genetic testing in age, clinical history and outcomes. PMD patients, in whom there is often a previous history of severe epilepsy and multisystemic involvement, showed distinct radiological features, such as chronic abnormalities, metabolic strokes and mitochondrial dysfunction on muscle biopsy. In contrast, cNORSE patients were younger (28.3 vs. 37.7 years), neurologically healthy, and lacked these features. The prognosis for PMD‐related RSE was worse, with all PMD cases deceased compared to 2/10 NORSE patients. Given these findings, the likelihood of an undiagnosed IMD in previously healthy adults presenting with NORSE is extremely low, especially with normal imaging.
PMD genetic tests in our tertiary mitochondrial centre follow a stepwise testing strategy, beginning with common mtDNA mutations, mitochondrial nuclear‐encoded gene panels and advancing to next generation sequencing (NGS) if necessary [14]. Eventually, in the ICU, the rapid exome could be adopted, to rapidly identify potentially treatable metabolic conditions [15]. Muscle tissue analysis should be reserved for cases needing verification of genetic findings, considering advancements in NGS and the limited sensitivity of traditional histopathology [14]. A proposed flowchart of investigations in cNORSE is provided in Figure 1.
Suggested flowchart of metabolic investigations in NORSE. aIt includes m.3243A>G in MT‐TL1, m.8344A>G in MT‐TK, and m.8993T>G/C in MT‐ATP6. bIncludes ABAT, AFG3L2, DGUOK, DNA2, DNM2, FBXL4, MFN2, MGME1, MPV17, OPA1, POLG, POLG2, RNASEH1, RRM2B, SLC25A4, SPG7, SUCLA2, SUCLG1, TFAM, TK2, TOP3A, TWNK, TYMP. LDH = lactate dehydrogenase, MRS = magnetic resonance spectroscopy, POLG = polymerase subunit gamma, RCEA = respiratory chain enzyme analysis, VLCFA = very long chain fatty acids, VUS = variant of unknown significance.
This study has limitations, including the potential for sampling bias due to the specialised nature of our ICU, retrospective design, small sample size and advancements in genetic testing over the study period. These issues highlight the need for collaborative research across specialist centers to advance diagnostic understanding in NORSE.
In conclusion, while identifying the cause of NORSE remains a significant challenge, our data suggest that IMD may be less likely in the absence of other suggestive clinical, radiological features, and/or positive family history. Therefore, exploring underlying metabolic diatheses should be clinically indicated, targeted and based on a clear rationale to avoid delays in diagnosis and appropriate management.
Author Contributions
Jennifer Kilmer: writing – original draft, methodology, visualization, formal analysis, data curation. George Ransley: methodology, investigation, writing – review and editing. Elaine Murphy: conceptualization, writing – review and editing, methodology. Michael G. Hanna: conceptualization, methodology, writing – review and editing, supervision. Robert D. S. Pitceathly: conceptualization, writing – review and editing, methodology, supervision, data curation. Sanjeev Rajakulendran: conceptualization, investigation, writing – review and editing, methodology, validation, supervision. Chiara Pizzamiglio: conceptualization, investigation, writing – review and editing, methodology, validation, data curation, supervision.
Ethics Statement
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1975, as revised in 2000. As the study includes only previously collected and available, non‐identifiable information, the UK Health Research Authority (HRA) was consulted and advised that it did not require review by an NHS Research Ethics Committee (REC).
Conflicts of Interest
The authors declare no conflicts of interest.
Supporting information
Data S1.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1L. J. Hirsch , N. Gaspard , A. van Baalen , et al., “Proposed Consensus Definitions for New‐Onset Refractory Status Epilepticus (NORSE), Febrile Infection‐Related Epilepsy Syndrome (FIRES), and Related Conditions,” Epilepsia 59, no. 4 (2018): 739–744, 10.1111/epi.14016.29399791 · doi ↗ · pubmed ↗
- 2N. Gaspard , B. P. Foreman , V. Alvarez , et al., “New‐Onset Refractory Status Epilepticus: Etiology, Clinical Features, and Outcome,” Neurology 85, no. 18 (2015): 1604–1613, 10.1212/WNL.0000000000001940.26296517 PMC 4642147 · doi ↗ · pubmed ↗
- 3M. Jindal , A. Neligan , and S. Rajakulendran , “Early and Established Status Epilepticus: The Impact of Timing of Intervention, Treatment Escalation and Dosing on Outcome,” Seizure 111 (2023): 98–102, 10.1016/j.seizure.2023.07.022.37556986 · doi ↗ · pubmed ↗
- 4B. Tumiene , C. R. Ferreira , and C. D. M. van Karnebeek , “2022 Overview of Metabolic Epilepsies,” Genes (Basel) 13, no. 3 (2022): 508, 10.3390/genes 13030508.35328062 PMC 8952328 · doi ↗ · pubmed ↗
- 5R. Y. Tan , A. Neligan , and S. D. Shorvon , “The Uncommon Causes of Status Epilepticus: A Systematic Review,” Epilepsy Research 91, no. 2‐3 (2010): 111–122, 10.1016/j.eplepsyres.2010.07.015.20709500 · doi ↗ · pubmed ↗
- 6A. Neligan , B. Kerin , M. C. Walker , and S. Rajakulendran , “New‐Onset Refractory Status Epilepticus (NORSE): The Queen Square Neuro‐ICU Experience,” Epilepsy & Behavior 125 (2021): 108387, 10.1016/j.yebeh.2021.108387.34781063 · doi ↗ · pubmed ↗
- 7J. Rankin , “Cerebral Vascular Accidents in Patients Over the Age of 60. II. Prognosis,” Scottish Medical Journal 2, no. 5 (1957): 200–215, 10.1177/003693305700200504.13432835 · doi ↗ · pubmed ↗
- 8M. Almannai , R. A. Al Mahmoud , M. Mekki , et al., “Metabolic Seizures,” Frontiers in Neurology 12 (2021): 640371, 10.3389/fneur.2021.640371.34295297 PMC 8290068 · doi ↗ · pubmed ↗