KIDINS220 Variant Associated With Hypoplasia of the Corpus Callosum and Aqueduct Stenosis
Kimia Ghannad‐Zadeh, Patrick Shannon, Rebekah Jobling, Elka Miller, Karen Chong, Erin Mathews, David Chitayat, Shiri Shinar

TL;DR
A new genetic variant in KIDINS220 is linked to brain development issues detected before birth.
Contribution
The first prenatal case of KIDINS220 variant associated with specific brain abnormalities is reported.
Findings
A novel de novo KIDINS220 variant was identified prenatally.
The variant was associated with ventriculomegaly, aqueductal stenosis, and corpus callosum hypoplasia.
Trio WES/WGS proved valuable for prenatal diagnosis and counseling.
Abstract
KIDINS220 plays a key role in neuronal survival, differentiation, and synaptic function. Abnormalities in its expression have been linked postnatally to neurodevelopmental disorders and SINO syndrome though prenatal presentations are rarely described. We report a novel de novo heterozygous KIDINS220 variant identified prenatally associated with bilateral ventriculomegaly, abnormal anterior horns, aqueductal stenosis, and a hypoplastic corpus callosum. This is the first prenatal case of such findings in KIDINS220, emphasizing the value of trio WES/WGS for diagnosis and counseling.
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
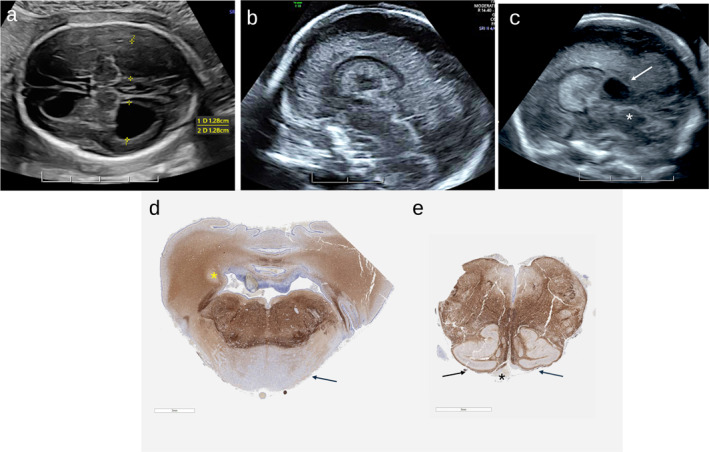 FIGURE 1
FIGURE 1| Case | Parental details | Gestation at diagnosis | Phenotypes (HPO terms) | Obstetric history | Family history | Outcome | ||
|---|---|---|---|---|---|---|---|---|
| 1 | Maternal | Age | 33 | 20 + 5 weeks |
Mesomelia (HP:0003027) Macrocephaly (HP:0000256) Brachycephaly (HP:0000248) Frontal bossing ( Flat face (HP:0012368) Thickened nuchal skin fold ( Aplasia of the fingers (HP:0009380) Aplasia of the toes (HP:0001991) Hemivertebrae (HP:0002937) Micropenis (HP:0000054) |
G3P0020 Two first trimester miscarriages | Nil | Termination of pregnancy |
| Ethnicity | English | |||||||
| Paternal | Age | 33 | ||||||
| Ethnicity | Sepharadic‐Jewish | |||||||
| Gene transcript | Mode of inheritance, gene OMIM | DNA variants, predicted effects, zygosity | ClinVar ID | Highest allele frequency in a gnomAD population | Interpretation |
|---|---|---|---|---|---|
|
| AD, 120130 | c.1536 + 1G > A, GT donor, heterozygous (de novo) | 3061119 | Not present |
Pathogenic PVS1 PS2 PM2 |
|
| AD, AR, 615759, 617296, 619501 | c.3899del; p.Pro1300Argfs*34, heterozygous (de novo) | 3349692 | Not present |
Pathogenic PVS1 PS2 PM2 |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsFetal and Pediatric Neurological Disorders · RNA modifications and cancer · Neonatal and fetal brain pathology
Summary
- What's already known about this topic?
- ◦Aberrant expression of KIDINS220 has been associated postnatally with neurodevelopment abnormalities and SINO syndrome.
- ◦Most individuals with SINO syndrome present postnatally with macrocephaly and ventriculomegaly. Prenatal presentation and neuropathological findings are rarely reported.
- What does this study add?
- ◦We report a case with hypoplasia of the corpus callosum, aqueduct stenosis and dilation of the anterior horns secondary to a de novo likely pathogenic heterozygous variant in KIDINS220.
- ◦To the best of our knowledge, this is the first prenatal report of these brain anomalies associated with the KIDINS220 variant.
Fetal Phenotype
1
The mother was a 33 years old primigravida and the father were of the same age. The couple was healthy and non‐consanguineous, and their family history was noncontributory. The conception was natural and resulted in dichorionic diamniotic twins with a vanishing twin in the first trimester. A detailed anatomical survey at 19 weeks showed bilateral ventriculomegaly of 14 mm and query absence of the cavum septi pellucidi (CSP). No extra CNS anomalies were seen. A neurosonogram at 20 + 5 weeks showed an appropriate gestational age male with bilateral moderate ventriculomegaly (13 mm, HP:0002119) with marked ballooning of the anterior horns and mild prominence of the third ventricle measuring 3 mm (Figure 1a). No fluid was seen in the aqueduct, raising suspicion for aqueduct stenosis (HP:0002410). There was a single choroid plexus cyst (HP:0011464). The CSP was thinned and compressed with a diffusely hypoplastic corpus callosum (HP:0002079 Figure 1b). The parieto‐occipital sulci were mildly effaced secondary to the ventriculomegaly and sulcation was otherwise age appropriate. There was bilateral thinning of the occipital parenchyma (HP:0002450, Tables 1A and 1B). Amniocentesis and MRI declined.
(a) Ultrasound images at 20 + 5 weeks of gestation axial plane demonstrating moderate bilateral ventriculomegaly (12.8 mm) and marked dilation of the anterior horns (arrows) as well as cortical thinning posteriorly. Midsagittal plane with diffusely hypoplastic corpus callosum. (b, c) Dilation of posterior recess of third ventricle (white arrow) secondary to aqueduct stenosis (white asterix). (d) Brainstem sections, stained with antibodies to Neurofilament Light Chain demonstrating demonstrating relatively small cerebellum, basis pontis (arrow) and small, poorly demarcated dentate nuclei (asterix). (e) Medulla with absent pyramids (asterix) and dysplastic inferior olivary nuclei (arrows).
Diagnostic Method
2
NIPT at 10 weeks gestation showed low risk for Trisomy 21, 13, 18, and sex chromosome aneuploidy in dizygotic male twins.
Early detailed ultrasound at 12 + 1 weeks showed the demise of one of the twins and the NT of the remaining twin was 1.1 mm.
Microarray analysis done on fetal banked DNA (fDNA) obtained at autopsy using an Illumina CytoSNP‐850K array showed a male fetus with no copy number gains or losses greater than 10 Kb across the genome.
Diagnostic Results and Interpretation
3
Trio whole‐exome sequencing performed on fDNA showed two likely pathogenic de novo sequence variants, one in KIDINS220 (c.3899del; p. Pro1300Argfs * 34) predicted to result in a frameshift, and a sequence variant in COL4A1 (c.1536 + 1G > A, GT donor) predicted to disrupt the GT donor site and interfere with normal splicing (Tables 1A and 1B). The couple was counseled that since these variants had likely arisen de novo, the empiric recurrence risk was approximately 1% due to the possibility of germline mosaicism.
Pregnancy Outcome and Autopsy Findings
4
The couple decided to terminate the pregnancy and the patient underwent an induction of labor and uncomplicated vaginal delivery at 21 + 1 weeks. Autopsy showed mainly brain anomalies including hypoplasia of the corpus callosum, mild aqueduct stenosis, severe hypoplasia of the corticofugal tracts and hypoplasia of the rhombic lip derivatives (Figure 1c,d).
Discussion
5
KIDINS220 codes for a protein preferentially expressed in the nervous system that controls neuronal cell survival, axonal and dendritic differentiation, and synaptic plasticity. Aberrant expressions of KIDINS220 have been associated with various neuropsychiatric and neurodegenerative diseases. Early termination and heterozygous loss‐of‐function variants in the last two exons of this gene are associated with pastic paraplegia, intellectual disability, nystagmus, and obesity (SINO syndrome) [1]. Other reported functional abnormalities include behavioral abnormalities, autism spectrum disorder, and attention deficit/hyperactivity disorder.
The effects of KIDIND220 variants on neurodevelopment are further characterized in mouse models. KIDINS220 knockout mice are not viable, with extensive deficits in dendritic growth, high rates of neuronal cell death, and significant ventriculomegaly [2]. Heterozygous KIDINS220 ^+/−^ mice are viable and do not show any major behavioral phenotype [2].
Biallelic KIDS220 likely pathogenic/pathogenic (LP/P) variants have been reported prenatally in three cases with the dominant early prenatal finding of ventriculomegaly [3, 4, 5]. In a recent report, Miremberg et al. reported a novel heterozygous variant in KIDINS220 presenting at 17 weeks with corpus callosum and brain stem dysgenesis, cortical malformation and hypertelorism [6]. The prenatal findings in our case are unique, consisting of ventriculomegaly, hypoplasia of the corpus callosum, and aqueduct stenosis on US and agenesis of the corticofugal tracts and hypoplasia of the rhombic lip derivatives on histopathology. These findings have yet to be described prenatally for this variant.
The variant reported here was heterozygous and resulted in frameshift and premature protein termination in exon 29 of the KIDINS220 gene. To the best of our knowledge, this variant has not been reported in the literature or in large population databases. However, due to its location in exon 29, it is expected to be pathogenic for autosomal dominant and recessive KIDINS220 associated disorders. The fetus reported here was also heterozygous for a de novo, likely pathogenic variant in COL4A1. In our reported case, there were no anomalies observed in neuromuscular or cardiac development, and there were no signs of intracranial hemorrhage, phenotypes characteristic of pathogenic COL4A1 variants.
In summary, we report a new LP de novo variant in KIDINS220 associated with aqueduct stenosis that would have likely resulted in SINO syndrome had the pregnancy continued. Our case highlights the importance of dedicated neurosonographic assessment of fetuses with ventriculomegaly as well as the importance of trio WES/WGS for accurate diagnosis and prenatal counseling.
Ethics Statement
The authors have nothing to report.
Consent
The parents consented for publication.
Conflicts of Interest
The authors declare no conflicts of interest.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1D. J. Josifova , G. R. Monroe , F. Tessadori , et al., “Heterozygous KIDINS 220/ARMS Nonsense Variants Cause Spastic Paraplegia, Intellectual Disability, Nystagmus, and Obesity,” Human Molecular Genetics 25, no. 11 (June 1 2016): 2158–2167. [Internet]. [Cited 2024 Sep 1], 10.1093/hmg/ddw 082.27005418 · doi ↗ · pubmed ↗
- 2F. Cesca , A. Yabe , B. Spencer‐Dene , et al., “ KIDINS 220/ARMS Mediates the Integration of the Neurotrophin and VEGF Pathways in the Vascular and Nervous Systems,” Cell Death & Differentiation 19, no. 2 (February 2012): 194–208. [Internet]. [Cited 2024 Sep 13], 10.1038/cdd.2011.141.22048155 PMC 3263493 · doi ↗ · pubmed ↗
- 3S. H. El‐Dessouky , M. Y. Issa , M. M. Aboulghar , et al., “Prenatal Delineation of a Distinct Lethal Fetal Syndrome Caused by a Homozygous Truncating KIDINS 220 Variant,” American Journal of Medical Genetics, Part A 182, no. 12 (December 1 2020): 2867–2876. [Internet]. [Cited 2024 Sep 1], https://pubmed‐ncbi‐nlm‐nih‐gov.myaccess.library.utoronto.ca/32909676/.32909676 10.1002/ajmg.a.61858 · doi ↗ · pubmed ↗
- 4I. L. Mero , H. H. Mørk , Y. Sheng , et al., “Homozygous KIDINS 220 Loss‐of‐Function Variants in Fetuses With Cerebral Ventriculomegaly and Limb Contractures,” Human Molecular Genetics 26, no. 19 (October 1 2017): 3792–3796. [Internet]. [Cited 2024 Sep 21], https://pubmed.ncbi.nlm.nih.gov/28934391/.28934391 10.1093/hmg/ddx 263 · doi ↗ · pubmed ↗
- 5V. Jacquemin , M. Antoine , S. Duerinckx , et al., “Trk A Mediates Effect of Novel KIDINS 220 Mutation in Human Brain Ventriculomegaly,” Human Molecular Genetics 29, no. 23 (December 1 2021): 3757–3764. [Internet]. [Cited 2024 Sep 1], 10.1093/hmg/ddaa 245.33205811 · doi ↗ · pubmed ↗
- 6H. Miremberg , R. Birnbaum , D. Trigubov , et al., “Prenatal Diagnosis of a KIDINS 220 De Novo Heterozygous Variant in a Fetus With a Complex CNS Anomaly,” Prenatal Diagnosis 44, no. 12 (2024): 1518–1521. [Internet]. [Cited 2024 Oct 9], 10.1002/pd.6682.39367534 · doi ↗ · pubmed ↗