Din Oversees Mesenchymal Stem Cell Homeostasis in Mouse Incisors
Xiaofang Wang, Changchun Dong, Bikash Lamichhane, Sanjaya Thapa, Yongxu Zhang, Shreyan Gupta, James J. Cai

TL;DR
The gene Din is crucial for maintaining mesenchymal stem cells in mouse incisors, affecting growth and healing.
Contribution
The study identifies Din as a key regulator of mesenchymal stem cell homeostasis in mouse incisors using a knockout model and single-cell RNA sequencing.
Findings
Din-deficient incisors showed arrested growth and impaired healing/regeneration after injury.
Din is essential for mesenchymal stem cells but not for epithelial stem cells or differentiated cells in incisors.
Din-deficient mesenchymal stem cells exhibited reduced stemness, motility, and osteogenesis potential.
Abstract
The murine incisor presents an excellent model for investigating stem cell homeostasis due to its regenerative capacity and continuous growth throughout the lifetime. Proper homeostasis of the dental epithelial stem cells (ESCs) and mesenchymal stem cells (MSCs) is pivotal for the continuous growth, tissue turnover and injury healing in murine incisors. By employing a newly developed knockout mouse model, we revealed that a predicted gene, Din (4930453N24Rik), plays pivotal roles in the homeostasis of MSCs in murine incisors. Din-deficient incisors exhibited arrested growth after eruption, and severely compromised healing/regeneration ability following injury. Although Din showed expression in multiple cell types in murine incisors, including both dental epithelium- and dental mesenchyme-derived naïve and differentiated cells, lineage-specific knockout of Din from epithelium, cranial…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
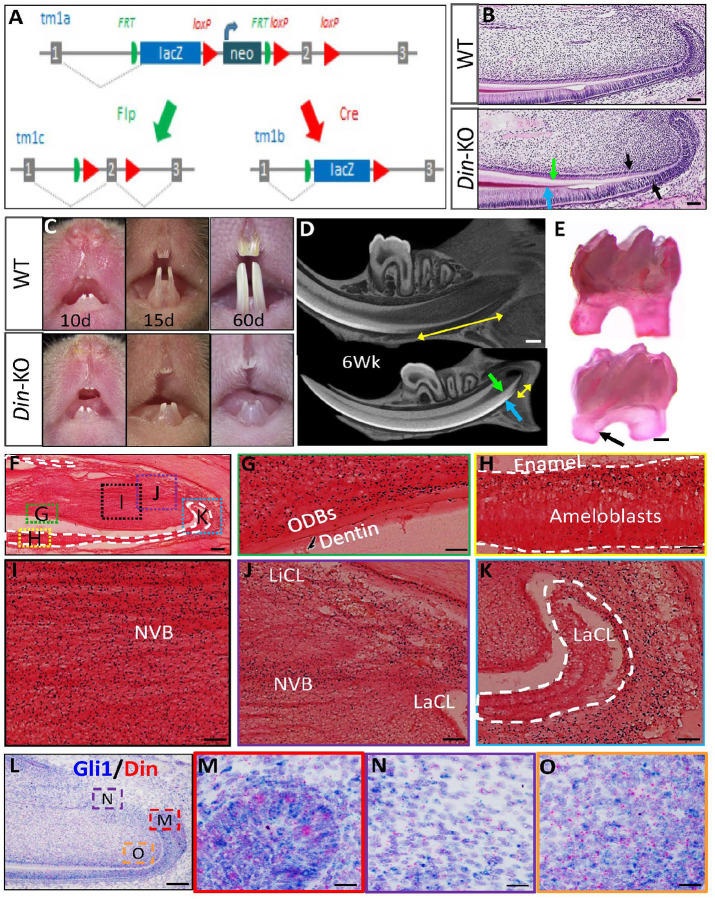 Figure 1
Figure 1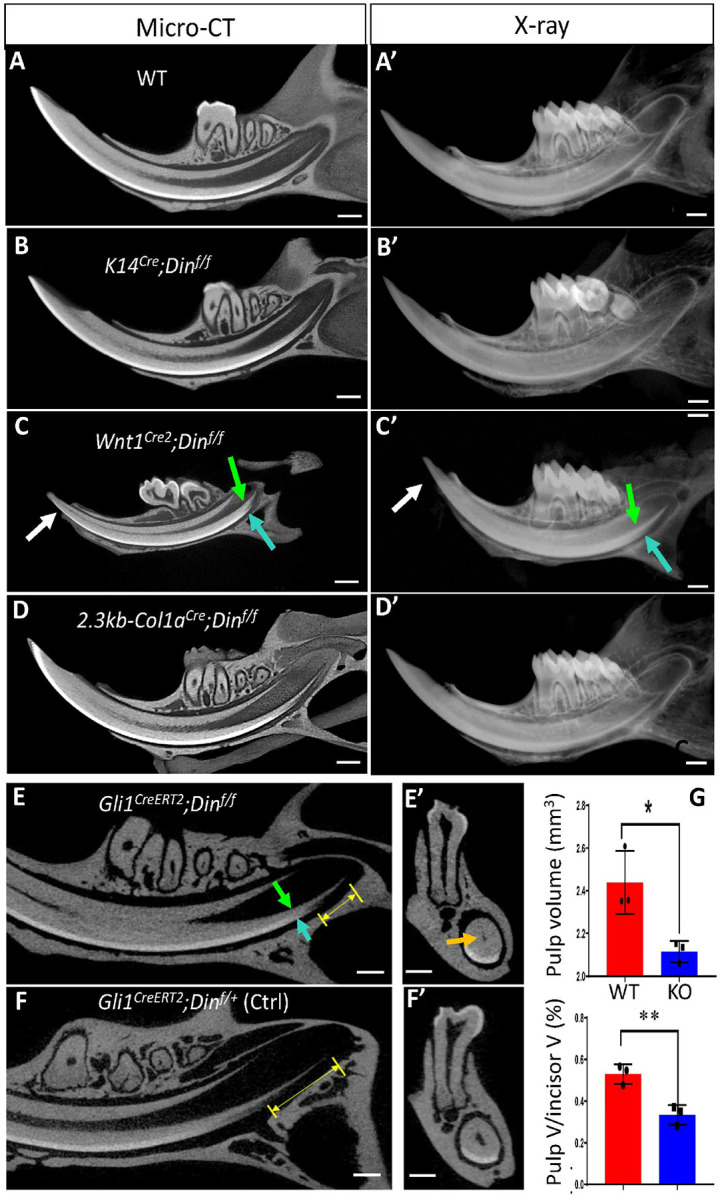 Figure 2
Figure 2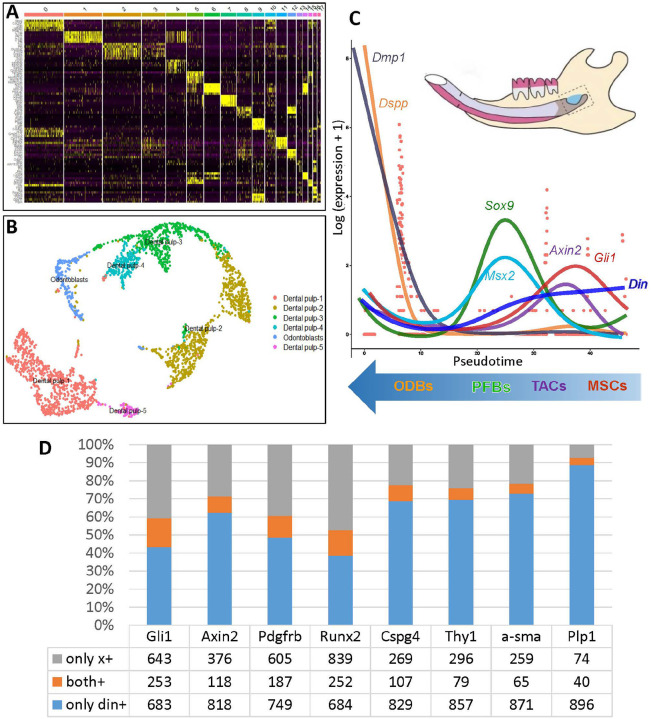 Figure 3
Figure 3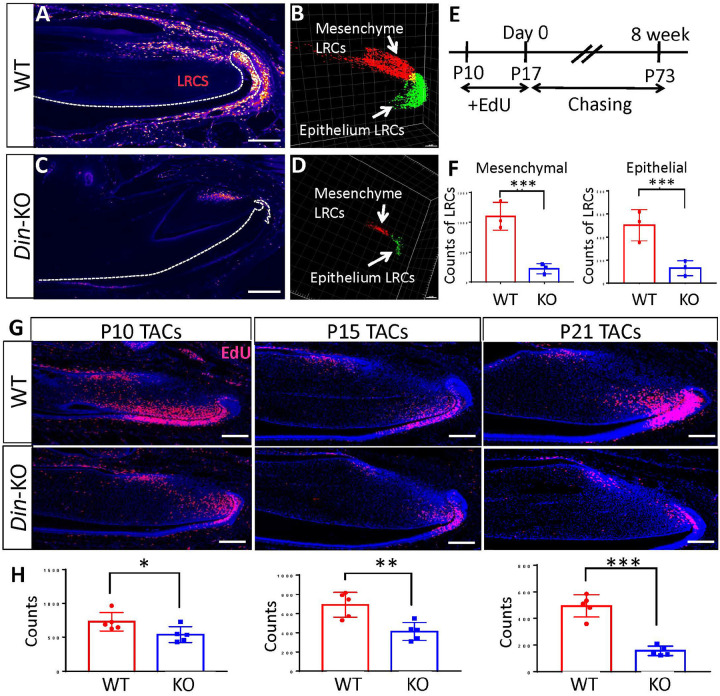 Figure 4
Figure 4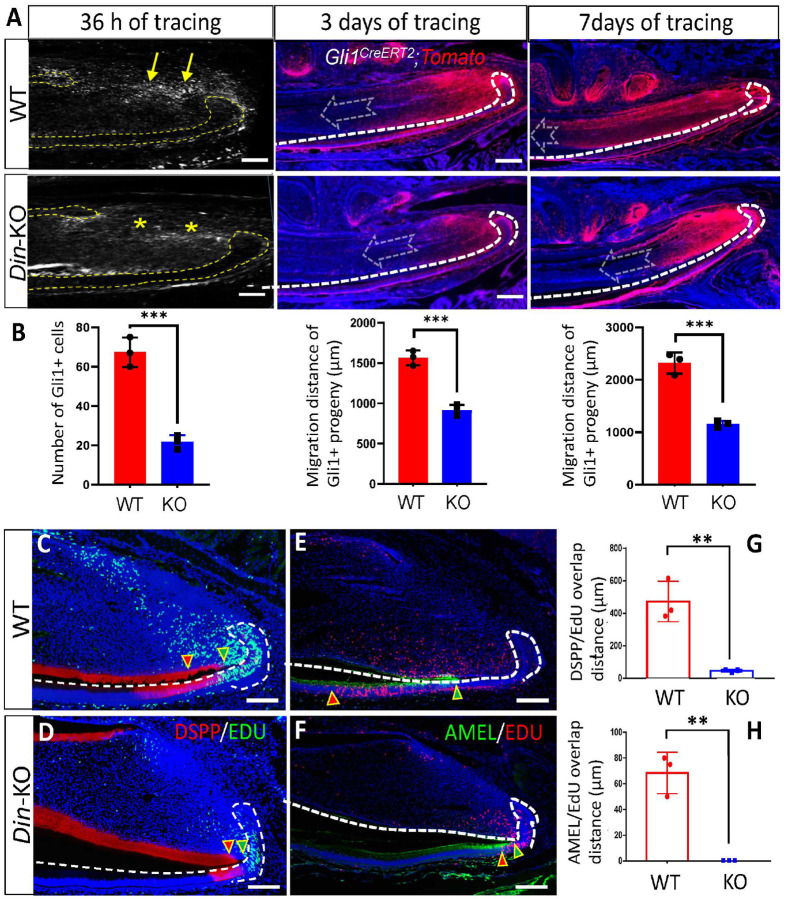 Figure 5
Figure 5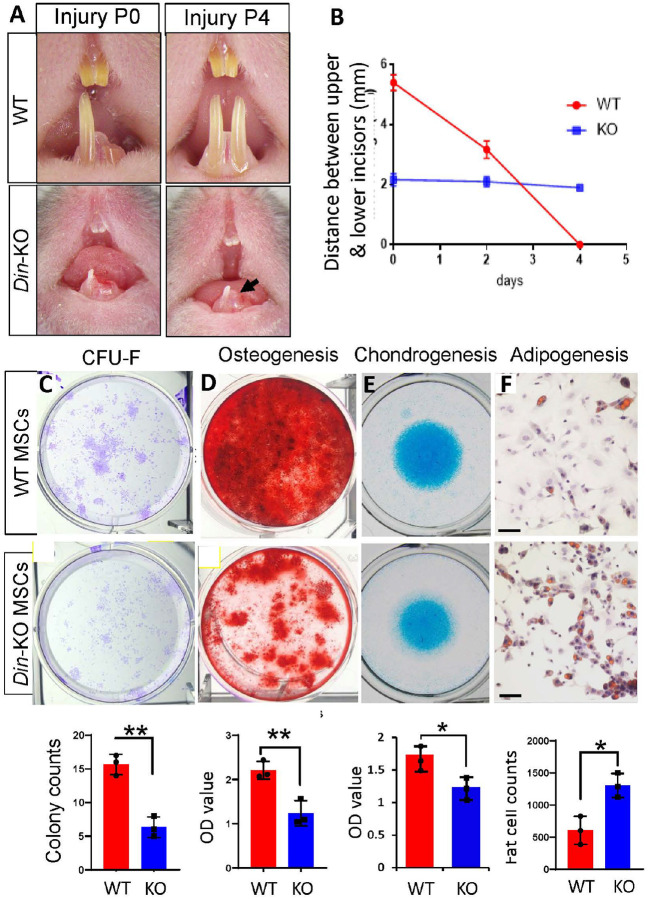 Figure 6
Figure 6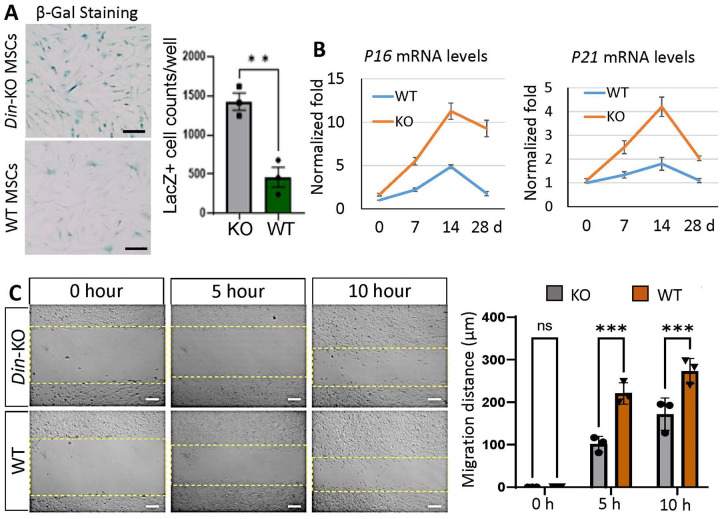 Figure 7
Figure 7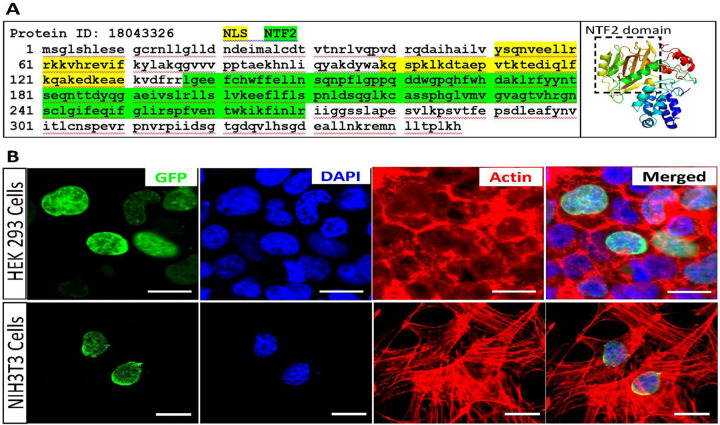 Figure 8
Figure 8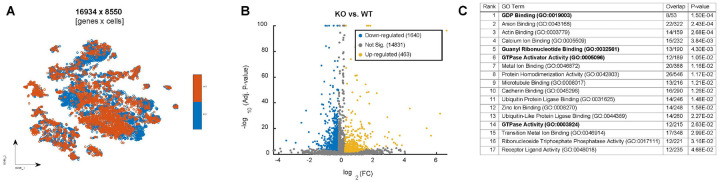 Figure 9
Figure 9Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsMesenchymal stem cell research · Neurogenesis and neuroplasticity mechanisms · Cancer Cells and Metastasis
INTRODUCTION
The mutation of Din gene arose in the Jackson Laboratory inbred C3H/HeJ strain mice in 1984^1^. The incisors of the mutant mice showed phenotypes in an autosomal recessive trait, including arrested growth after eruption, “overgrowth” of dentin, and a reduced pulp chamber, collectively referred to as Dense incisors or Din^1^. The causal gene for Din was initially mapped to chromosome 16 by linkage analyses^1^, and later identified as the predicted gene 4930453N24Rik via whole exome sequencing^2^. So far, little is known about the function of Din gene. The “overgrowth” of dentin due to arrested growth of incisors in Din mice, reminiscent of similar phenotypes observed in several mutant mice with dysregulated signaling in incisor stem cells or transit amplifying cells (TACs)^3–5^, which implies a potential role of Din in the homeostasis of murine incisors, particularly considering that mice experiencing issues with incisor renewal often exhibit similar phenotypes^3–7^.
Mice possess the unique ability to completely renew their incisors every 35 to 45 days. This trait, shared by all rodents, makes mice highly valuable for studying the regeneration of tissues through stem cells. Within a month, the epithelial and mesenchymal compartments of murine incisors replenish all their cells rapidly^8^. The self-renewal of incisor relies on quiescent epithelial stem cells (ESCs) in the cervical loop region^9, 10^ and mesenchymal stem cells (MSCs) localized near the cervical loops and neurovascular bundle that give rise to TACs^11, 12^. The ESCs and MSCs at the proximal side of incisor supply the progenitors that differentiate into ameloblasts and odontoblasts to form enamel and dentin continuously^3, 6, 13^. In response to injury or stress, the repair system of murine incisors gets activated which are backed by these stem cells that have intrinsic ability to self-renew and regenerate tissue-specific mature cells to replace damaged enamel, dentin and dental pulp. The tissue turnover and healing process require the continuous supply of cells that can divide over a period of time. Thus, the homeostasis of stem cells and their differentiation into the required tissue-specific cells are a must for proper turnover/healing which are governed by various signaling mechanisms^14^.
Stem cell homeostasis involves a series of tightly regulated cellular events, including population maintenance, cell cycle switching, directed migration, and guided differentiation, etc. Proper homeostasis of stem cells is crucial for postnatal growth, tissue turnover, and injury repair in both human and rodent teeth^3, 15^. Several regulatory mechanisms have been identified associated with the homeostasis of ESCs during enamel renewal/turnover in murine incisors^9, 16–19^. Due to the significant application prospects of MSCs in regenerative medicine/dentistry, there is a greater focus on research regarding MSC homeostasis in murine incisors, which is associated with tissue turnover and injury healing in dentin-pulp complex^3–5, 7, 12, 20–22^. These studies have revealed the regulatory mechanisms governing specific aspects of MSC homeostasis in mouse incisors. However, it remains unclear whether there are factors overseeing various aspects of MSC homeostasis and if a cohesive regulatory mechanism exists to coordinate the processes.
Small GTPases function as nodal points that integrate upstream regulatory inputs and disseminate effector outputs to modulate extensive signaling pathways and cytoskeleton reorganization to regulate cell proliferation, differentiation, polarization and motility, etc. They make binary on/off decisions through controlled loading of GTP (activation) and hydrolysis of GTP to GDP (inactivation). The cellular regulation of this cycle involves guanine nucleotide exchange factors (GEFs) which accelerate intrinsic GDP/GTP exchange, GTPase-activating proteins (GAPs) which terminate the signaling event, and guanine nucleotide dissociation inhibitors (GDIs) which bind to prenylated GTPases and control their cycle between the cytosol and membrane^23^.
It has been shown that small GTPases regulate the homeostasis and cell fate of several stem cell lineages^24–34^, and are implicated in tooth development and tissue turnover. For instance, integrin α3 regulates TAC proliferation via FAK and Cdc42, driving tissue renewal in mouse incisors^35^; RhoA, Rac1, and ROCK are associated with ameloblast differentiation^36^; and CDC42-mediated Wnt signaling promotes odontogenic differentiation of dental papilla cells during tooth root elongation^37^.
In this study, we demonstrate that Din is essential for MSC homeostasis in murine incisors, but is dispensable for odontoblasts and dental epithelium-derived cells, such as ameloblasts and ESCs. Din oversees multiple aspects of MSC homeostasis, including stemness, proliferation, differentiation, migration, and population maintenance. These functions are likely mediated through the binary molecular switches of Rho GTPases.
RESULTS
Din is Essential for the Postnatal Growth and Renewal of Murine Incisors.
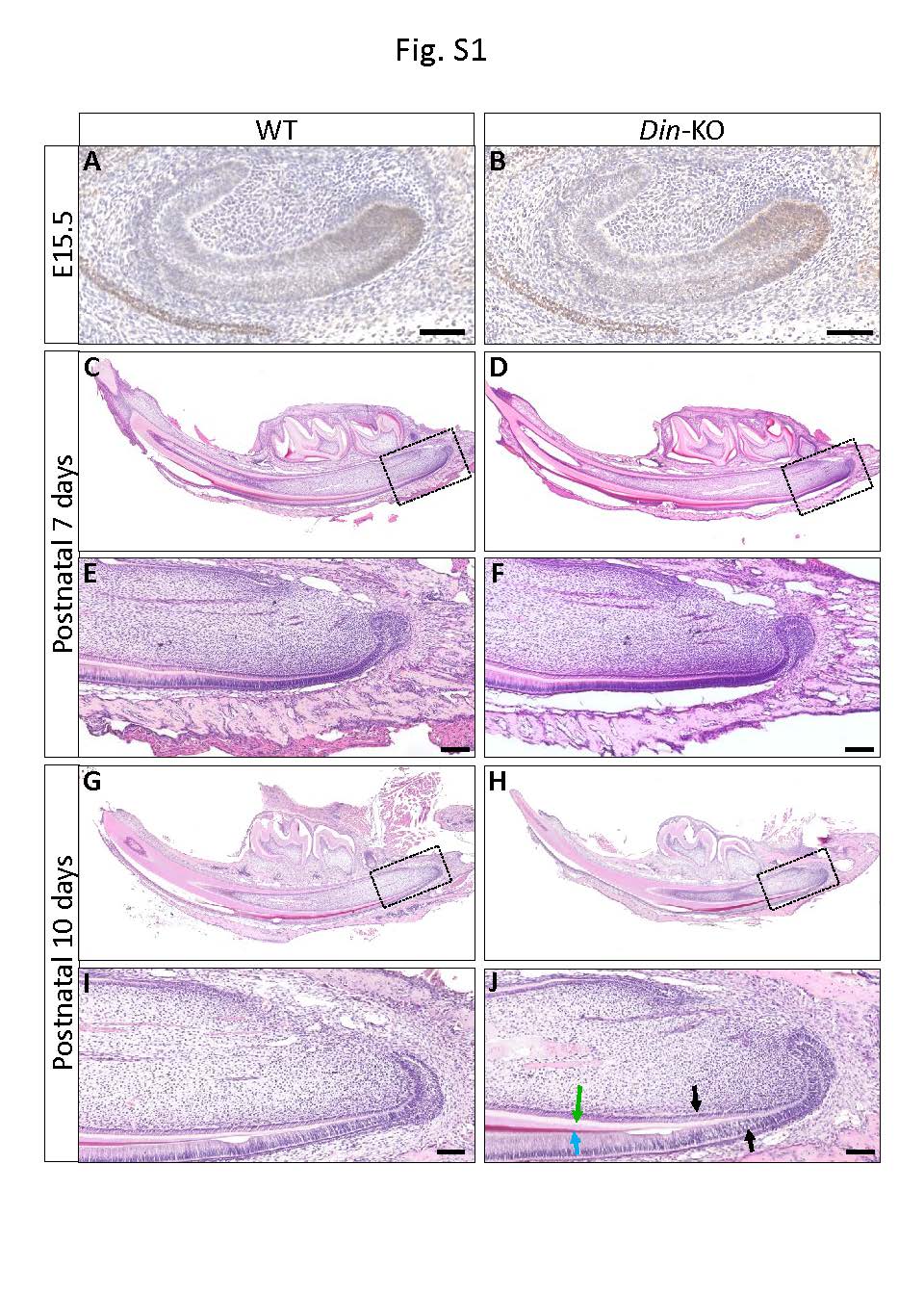
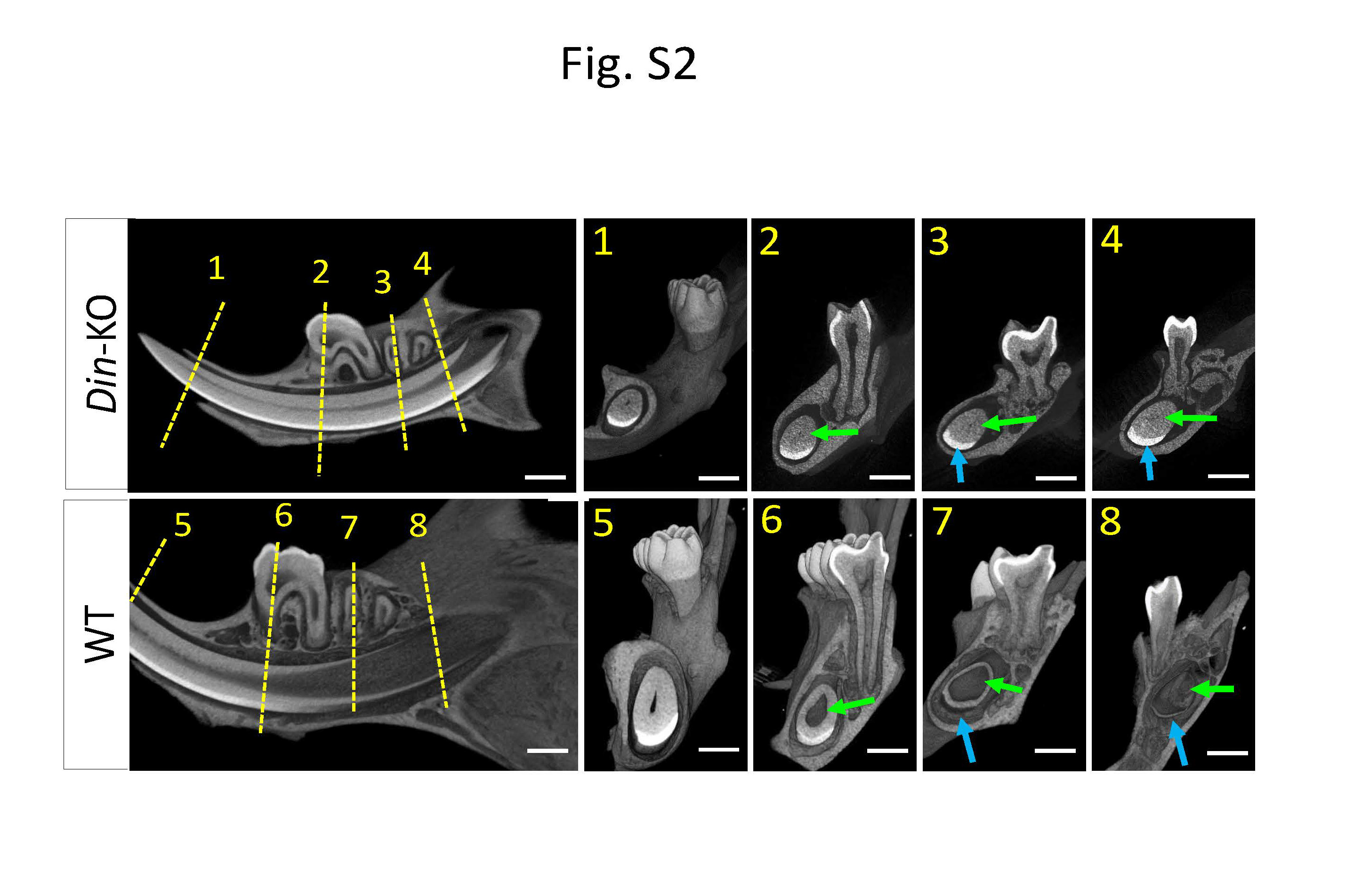
Din deficiency did not affect the development of embryonic teeth but significantly compromised the postnatal growth and renewal of murine incisors and root development of molars. Histology of embryonic and postnatal incisors did not identify differences between Din-KO and WT until postnatal 10 days (Fig. S1). Din-KO incisorscan normally erupt into oral cavity. Subsequently, they started showing extremely slow or arrested growth (Fig. 1B and C), while their dentin continued accumulating until the pulp chamber was obstructed at older age (Fig. 1D, Fig. S2). Mature dentin and enamel were observed at the proximal side near cervical loops where normally they should not be present (Fig. 1D, Fig. S2). The molars of Din-KO mice showed a largely normal crown, with compromised postnatal growth of dental roots (Fig. 1E). The presecretory and secretory ameloblasts and odontoblasts in P10 Din-KO incisors displayed more mature and higher column morphology than WT (Fig. 1B), and laid down more dentin and enamel matrix at the proximal side near the cervical loops (Fig. 1B and D). No differences were identified in the teeth between male and female Din-KO mice.
The Expression Pattern of Din in Murine Incisors.
The Din-KO allele had a LacZ reporter driven by endogenous Din promoter, which can be used to indicate Din expression (Fig. 1A). X-Gal staining on sagittal cryosections of lower incisors from 4-week-old Din-KO heterozygotes (normal mice) showed that Din is expressed in both differentiated and undifferentiated dental cells, including pre- and mature odontoblasts and ameloblasts (Fig. 1F–H), and dental pulp cells in the proximal neurovascular bundle (NVB) and between the labial and lingual cervical loops, where the putative dental MSC niches reside (Fig. 1I and J). Labial cervical loop (LaCL) only showed sparse expression, while the dental follicle cells posterior to LaCL displayed robust signals (Fig. 1K).Dual RNAScope staining of Din and Gli1 showed co-expression of the two transcripts in Gli1+ MSCs and TACs in murine incisors (Fig. 1L–O).
Din is Essential for Dentinogenesis but Dispensable for Amelogenesis in Murine Incisors.
The development of dentin and enamel involves reciprocal interactions between the dental epithelium and dental mesenchyme. Given that Din expression was detected in cells derived from both dental epithelium and dental mesenchyme, and that the dental defects observed in Din-KO mice incisors affected both dentin and enamel, there is a need to clarify whether Din functions in either dental tissue or both of them. To this end, Din-floxed mice were crossbred with K14-Cre and Wnt1-Cre2 transgenic mice to conditionally knock out (cKO) Din from either tissue. Surprisingly, the K14-Cre derived cKO mice did not show any dental abnormities, while the Wnt1-Cre2 derived cKO mice recapitulated the dentin and enamel phenotypes of Din-KO mice (Fig. 2A–C’). These results indicate that Din plays a critical role in dentinogenesis but is not essential for amelogenesis in murine incisors. The enamel defects observed in the incisors of Din-KO mice appear to be secondary effects resulting from defective dentinogenesis.
Din is Essent ial for MSCs but not Odontoblasts in Murine Incisors.
To determine the cell stages at which Din functions in dental mesenchyme-derived cells, we conditionally knocked out Din from cells expressing Col1a and MSCs expressing Gli1 using 2.3-kb Col1a1-Cre and Gli1-Cre^ERT2^. Surprisingly, Din-cKO from Col1a-expressing cells (odontoblasts and fibroblasts) did not result in any dental abnormities (Fig. 2D and D’), while Din-cKO from Gli1+ MSCs recapitulated a milder version of dentin and enamel defects similar to the Din-KO mice (Fig. 2E–G). These results collectively suggest that Din is dispensable for odontoblasts but essential for dental MSCs in murine incisors.
In addition to Gli1+ MSCs, there are multiple MSC subsets in mouse incisors responsible for tissue homeostasis and/or injury healing^38^. To determine Din expression in the cell populations in murine incisors, we performed scRNA-seq on the proximal tissues from the lower incisors of P15 WT mice. Out of the 17 cell groups identified by cell clustering (Fig. 3A), we focused on the 6 groups from mesenchyme, including dental pulp, dental follicle, and odontoblasts (Fig. 3B). Pseudo-time assay revealed a decline in Din expression levels along the trajectory of MSC differentiation, with highest levels in MSCs and TACs, followed by low levels in pulp fibroblasts (PFbs) and odontoblasts (ODbs) (Fig. 3C). The scRNA-seq data further demonstrated Din expression in multiple MSC subsets in murine incisors, but in each subset, only a fraction of MSCs express Din (Fig. 3D). These data collectively suggests that Din is essential for dental MSCs but dispensable for odontoblasts. The Din-KO dental defects may involve multiple MSC subsets.
Din is Essential for the Homeostasis of Dental MSCs During Postnatal Growth of Murine Incisors.
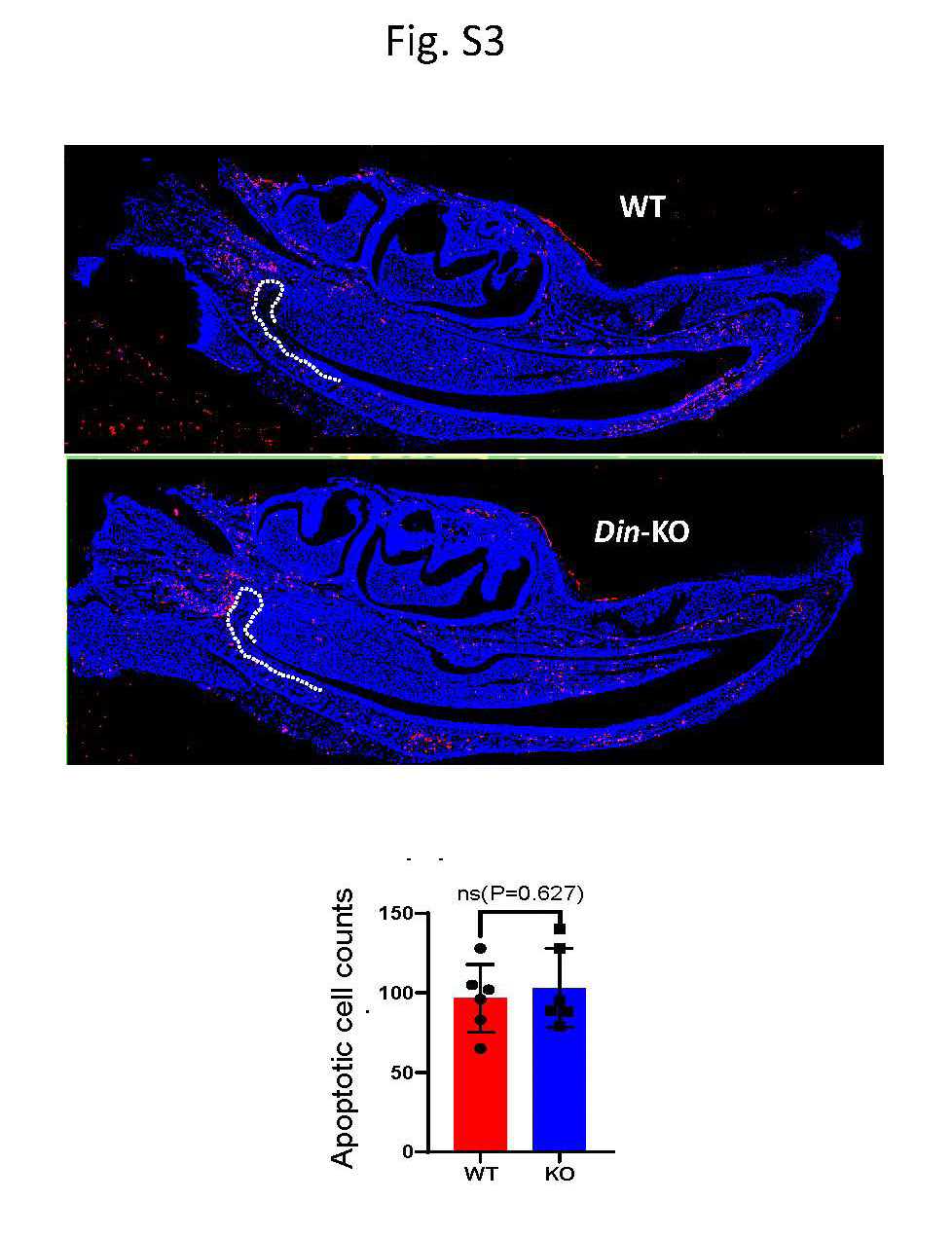
Mouse incisor is an excellent model for studying homeostasis of stem cells due to its rapid turnover during postnatal growth and regenerative capacity. To determine the impact of Din deficiency on the homeostasis of dental MSCs, we examined the label retaining cells (LRCs) in mouse incisors. Din-KO and WT mice were i.p. injected with EdU (50 mg/kg) at P10 for 7 consecutive days, and sacrificed after 8 weeks. The mandibles were processed for tissue clearing and whole-mount EdU staining followed by confocal microscopy and Imaris quantitation as previously described^39, 40^. The Din-KO incisors showed dramatically less LRCs in both MSC niche and cervical loops than WT (Fig. 4A–D, F). Next, we examined TACs proliferation in incisor at P10, P15 and P21. EdU (50 mg/kg) was i.p. injected 2 h prior to sacrifice. IF staining of DSPP and AMEL was conducted on sagittal cryosections of lower incisors. Din-KO incisors showed reduced TACs in dental mesenchyme and epithelium, starting at P10 and worsening with age (Fig. 4G and H). Despite UniProt and MGI gene annotations associating Din with “cell apoptosis,” we did not observe significant changes in TUNEL staining in Din-KO incisors. (Fig. S3).
To determine the impacts of Din deficiency on the homeostasis of MSC progenies in mouse incisors, we performed lineage tracing of Gli1+ MSCs. Gli1-CreERT2 and tdTomato transgenic mice were crossbred with Din-KO and WT mice. CreERT2 was induced by a single injection of tamoxifen (75 mg/kg, i.p.) at P11. The expression of tdtomato was traced for 36 h, 3 days, and 7 days. The Din-KO incisors showed significantly less Gli1+ MSCs than WT at 36 h, and their progeny cells displayed dramatically less/slower contribution to the tissue turnover from proximal toward incisal side at 3 and 7 days of tracing (Fig. 5A and B). IF staining detected robust expression of DSPP and AMEL on the proximal side near cervical loops in Din-KO incisors (Fig. 5C–F). The overlapping distance of DSPP/EdU or AMEL/EdU, indicative of the differentiation potential of TACs, was dramatically diminished in Din-KO incisors (Fig. 5C, E and G).These data strongly suggest that Din is essential for MSCs homeostasis in murine incisors during the postnatal growth and tissue turnover.
Din is Essential for the Homeostasis of Dental MSCs During Injury Healing/Regeneration of Murine Incisors.
The MSCs responsible for tissue homeostasis and injury repair are often attributed to different subpopulations^3, 11, 21, 38^. Our scRNA-seq and open-access data showed Din expression in multiple MSC subsets in murine incisors (Fig. 3D). To determine if Din is essential for the MSCs subsets responsible for injury healing, we conducted injury repair experiments on the lower incisors in 6-week-old mice^7, 41^. The broken incisors of WT mice regenerated to normal length in 5 days, while Din-KO incisors lacked regenerative healing (Fig. 6A and B). This data suggests that Din is essential for the homeostasis of MSC subsets responsible for injury healing and regeneration in murine teeth.
Din Oversees Multiple Aspects of MSCs Homeostasis.
To investigate whether Din-KO MSCs undergo cellular behavior/capability changes during tissue turnover and injury repair, we isolated Gli1+ MSCs from mouse incisors for CFU-F and tri-lineage differentiation assays. The Din-KO MSCs exhibited reduced capacities for CFU-F, osteogenesis, and chondrogenesis compared to normal controls (Fig. 6C–E), whereas their adipogenesis potential was significantly enhanced (Fig. 6F). Senescence assays, including β-Gal staining and Q-PCR analysis of senescence markers, unveiled a notable increase in aging cells and elevated expression levels of P16 and P21 in Din-KO MSCs (Fig. 7A and B). Scratch assays conducted on monolayer MSCs demonstrated a markedly impaired motility in Din-KO MSCs compared to normal controls. (Fig. 7C).
Din Regulate MSCs Homeostasis through Rho GTPases.
Using Phyre2 and cNLS Mapper analyses, we identified a Nuclear Transport Factor 2 (NTF2) domain and two Nuclear Localization Sequences (NLS) in the DIN protein. (Fig. 8A). NTF2 is a cytosolic protein mediating the import of GDP-bound small GTPase RAN (RAs-related Nuclear protein) from the cytoplasm into the nucleus, which is essential for the function of RAN in cargo receptor-mediated nucleocytoplasmic transport. The presence of NTF2 and NLS domains in DIN suggests that DIN may translocate into the cell nucleus to carry out its function. We determined the subcellular location of DIN by constructing and transfecting a pCMV6-Din-GFP mammalian expression vector into HEK-293 and NIH/3T3 cells. The DIN-GFP fusion proteins showed robust nuclear (membrane) localization in both cell lines (Fig. 8B).
To investigate the potential mechanism through which Din regulates the homeostasis of dental MSCs, we compared gene expression in fibroblasts between Din-KO and WT incisors. The processed scRNA-seq data contains 16,934 genes expression in 8,550 fibroblast cells: 4,478 from the Din-KO samples and 4,072 from the WT samples (Fig. 9A). Using differential expression analysis, we identified 1,640 down-regulated genes (with expression level is lower in KO samples) and 463 up-regulated genes (with expression level is higher in KO samples, Fig. 9B). GO enrichment analysis with down-regulated genes identified 17 significant molecular function terms with Enrichr p-value < 0.05 (Fig. 9C). These include GDP Binding (GO:0019003), Guanyl Ribonucleotide Binding (GO:0032561), GTPase Activator Activity (GO:0005096), and GTPase Activity (GO:0003924). These results are consistent with the anticipated function of the NTF2 domain. Associated GTPase-related genes include: Vav3, Arhgap10, Arhgap42, Acap2, Rgs3, Dlc1, Chn1, Arhgap29, Arhgap28, Iqgap2, Dnm1l, and Smap1, which were all significantly downregulated in the Din-KO fibroblast populations (Supplementary Table 1).
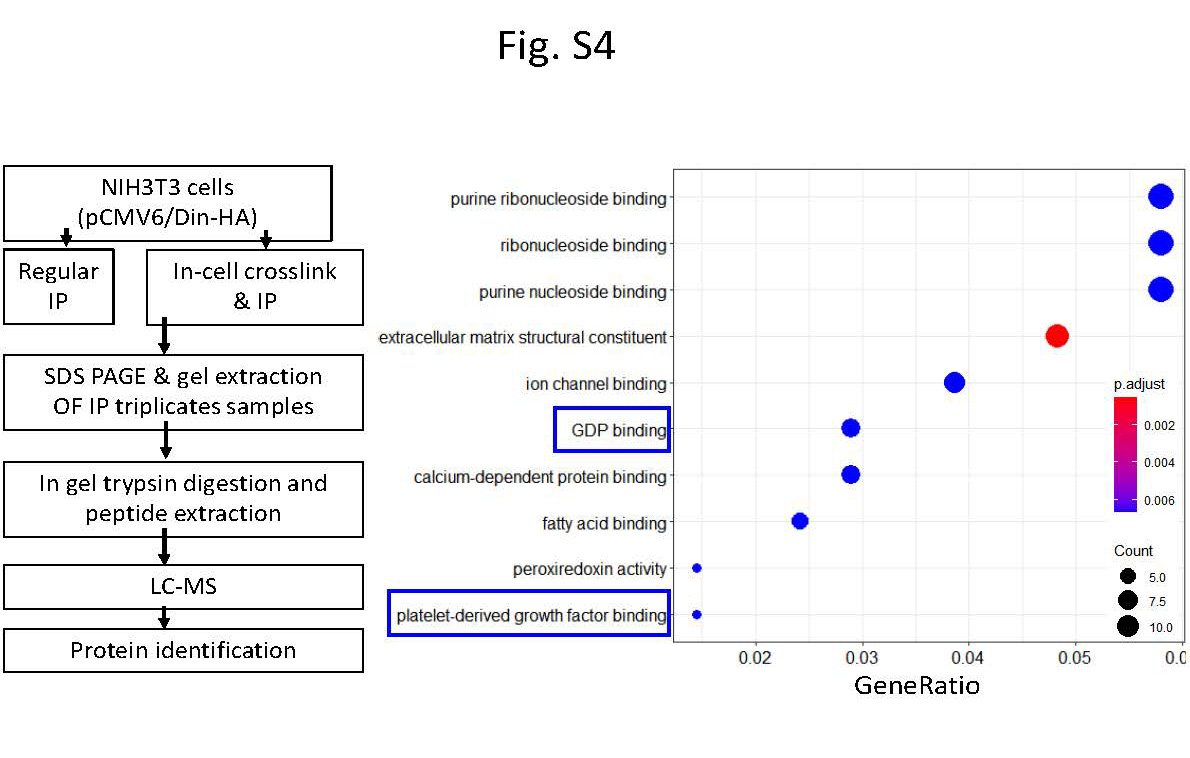
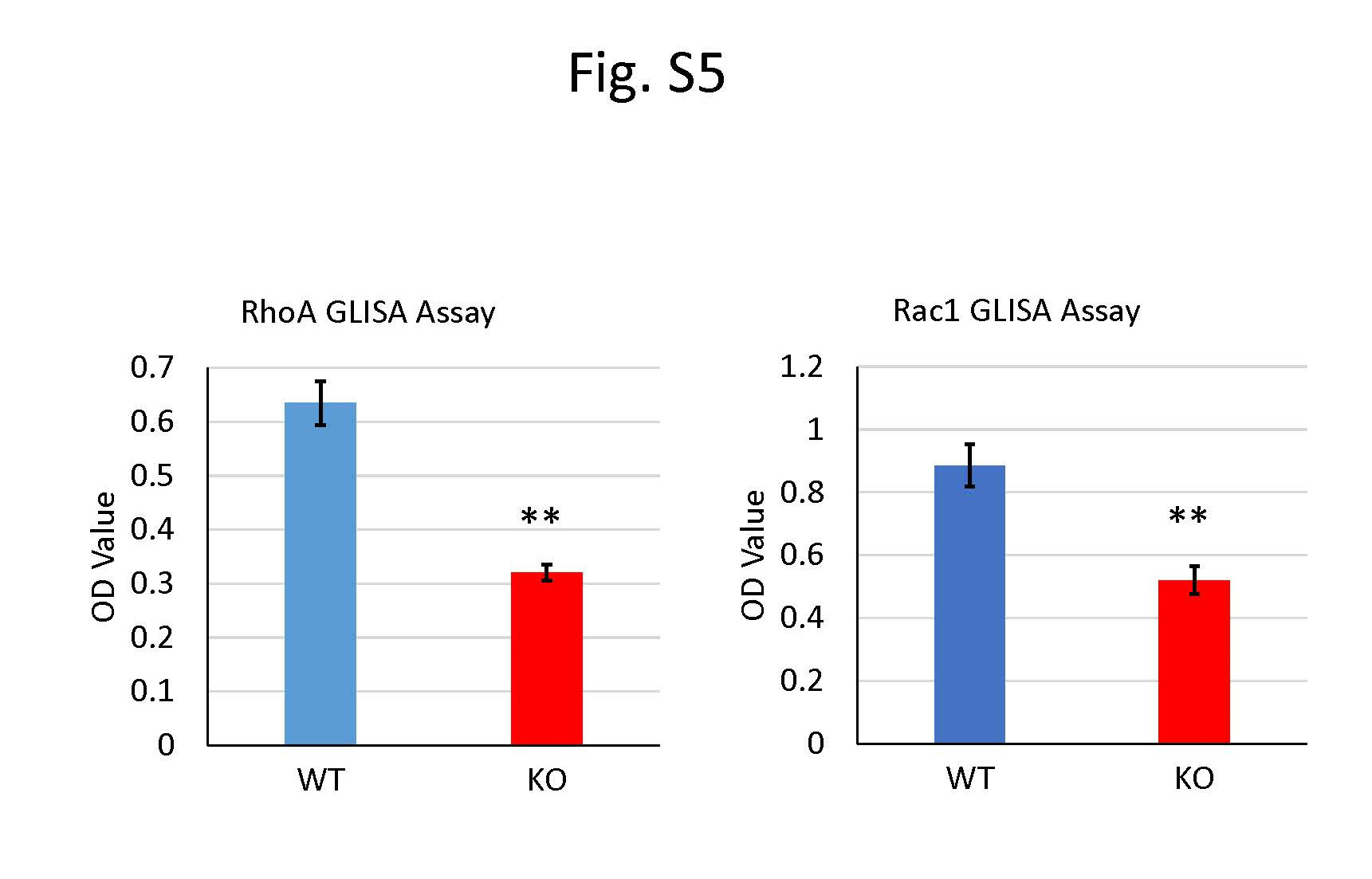
To identify small GTPases that potentially interact with DIN, we conducted Co-IP assays on lysates prepared from NIH/3T3 cells stably transfected with pCMV6-Din-HA constructs, followed by LC-MS (liquid chromatography - mass spectrometry) assays (Fig. S4). Four Rho GTPases, namely Nras, RhoA, Cdc42, and Rac1, were identified as potential interactors with DIN, as evidenced by their 3.4 – 82.5 fold higher abundance in the DIN-HA pulled-down immunoprecipitations (Supplementary Table 2). The activity of these GTPases was further examined in the Gli1+ MSCs isolated from murine incisors using the RhoA/Rac1/CDC42 G-LISA Activation Assay Bundle Kit and Ras G-LISA Activation Assay Kit. The Din-KO MSCs showed significantly lower activity (GTP-bound) levels of RhoA and Rac1 compared to WT MSCs (Fig. S5), without apparent alteration in the levels of active Nras and Cdc42. These results collectively suggest that Din may positively regulate RhoA and Rac1 in MSCs within murine incisors.
DISCUSSION
Din exhibits a broad expression pattern in both dental epithelial and mesenchymal naïve and differentiated cells. Correspondingly, Din-KO mice show defects in both dentin and enamel in their incisors. However, our lineage- and tissue-specific knockout studies targeting the epithelium, cranial neural crest, Col1a1-expressing cells, and Gli1+ MSCs demonstrated that Din is dispensable for odontoblasts and dental epithelium-derived cells (such as ameloblasts and ESCs) but is essential for dental MSCs in murine incisors. These findings indicate that the enamel defects observed in Din-KO mice are secondary to impaired dentinogenesis, and the primary dentin defects arise from abnormalities in MSCs but not from differentiated odontoblasts. Tooth development relies on proper epithelium-mesenchyme interactions. The epithelial or mesenchymal signaling can regulate the behavior of stem cells located in the counterpart compartment in murine incisors^6, 9, 42–45^. Thus, primary defects in the MSCs may have secondary effects on the ESCs and vice versa. In this study, both ESCs and MSCs in the Din-KO incisors exhibited a significant decrease in quantity, along with compromised proliferation and differentiation, suggesting that the potential interaction between MSCs and ESCs may be associated with the secondary enamel defects observed in the incisors of Din-KO mice.
Proper homeostasis of MSCs is crucial for the tissue turnover and injury repair in teeth^38^. Several studies have identified regulatory mechanisms governing certain aspects of MSC homeostasis in murine incisors^3–5, 7, 12, 20–22^. It remains unclear whether a cohesive regulatory mechanism oversees the homeostasis of MSCs. Through in vivo lineage tracing of MSCs in Din-KO and Din-cKO mice, along with in vitro characterization of the cellular potential and capacities of Din-deficient MSCs, we found that Din is essential for multiple aspects of MSC homeostasis, including maintaining stemness, migration, aging, proliferation, and differentiation. It plays a central role in regulating MSC homeostasis in murine incisors. This overarching effect is evident in two aspects: first, Din is expressed in multiple MSC subpopulations within murine incisors; second, Din may regulate MSCs through GTPases, which are binary molecular switches involved in the extensive regulation of various cellular processes. Through scRNA-seq and computational analysis, we identified a connection between Din and GTPases. Additionally, using IP, MS, and GLISA, we preliminarily identified several Rho GTPases that may interact with Din. Future studies will involve a series of in vivo and in vitro experiments to validate and further investigate the regulatory relationship between these molecules.
The expression of odontoblast and ameloblast differentiation markers, DSPP and AMEL, in the Din-KO incisors exhibited a ‘stronger’ and ‘more proximal’ pattern near the cervical loops, with reduced or absent overlap with EdU+ cells. It should be noted that this seemingly ‘stronger’ and ‘earlier differentiation’ pattern does not indicate enhanced proliferation and differentiation. On the contrary, it suggests a reduced capacity of the MSCs and ESCs regarding these aspects. The Din-deficient dental stem cells cannot supply adequate new TACs to support the turnover of ameloblasts and odontoblasts toward incisal side. Consequently, pre-odontoblasts and pre-ameloblasts differentiate into high-column mature cells in situ near the cervical loops, leading to robust DSPP and AMEL expression and the formation of mature enamel and dentin on the proximal side of the incisors.
The various MSC subpopulations in the dental pulp enable continuous growth and rapid repair of murine incisors. However, tissue turnover and injury repair are usually associated with different MSC subsets^3, 11, 21, 38^.For instance, Gli1+, Thy1+, and Plp1+ cells are slow-cycling cells regulating incisor tissue turnover and/or injury healing. αSMA+ and Cspg4+ MSCs function as pericytes involved in tissue healing following injury, whereas Pdgfrb+ and Axin2+ cells represent TACs derived from MSCs re-entering the cell cycle. Our scRNA-seq data showed that Din is expressed in most of the MSC subsets, and in each subset, only a fraction of MSCs express Din. It remains unclear whether the expression or absence of Din within the same subset leads to differences in MSCs behavior and cell fate. Future single-cell analysis and cellular characterizations after sorting the Din+ and Din− populations within each MSC subset and subset-specific knockouts may provide more valuable insights to clarify these questions. As a starting point, we knocked out Din from the largest MSC subpopulation in mouse incisors, the Gli1+ MSCs. The resulting conditional knockout incisors displayed a milder phenotype compared to the conventional knockout, which may be partially attributed to the efficiency of Cre, but more likely reflects Din’s role in multiple MSC subsets. Specifically, the knockout of Din in a single MSC subpopulation cannot fully recapitulate the knockout phenotype.
In summary, we have identified that a predictive gene Din plays central role in regulating multiple aspects of MSC homeostasis in murine incisors, which is essential for their continuous growth and injury repair. The mechanism underlying the regulation of incisor MSC homeostasis is associated with small GTPases.
MATERIALS & METHODS
Animals
All animal experiments were carried out according to the protocol approved by the Institutional Animal Care and Use Committee (IACUC) of Texas A&M University School of Dentistry (Dallas, TX, USA), and performed by the NIH Guide for the Care and Use of Laboratory Animals.
Generation of Din-KO1st, Din-KO and Din-flox Alleles
Din-KO^1st^ mice were generated using a gene-trap targeted ES cell clone 4930453N24Rik^tm1a(KOMP)Mbp^ from The Knockout Mouse Project (KOMP). The KO^1st^ allele (Fig. 1A, tm1a) having loxP and Frt sequences can be transformed into conventional Din-KO (Fig. 1A, tm1b) or Din^flox^ (Fig. 1A, tm1c) allele via breeding with germline expressing Cre or Flp transgenic mice.
Generation of Din Conditional Knockout (cKO) Mice
All Cre lines used for generating cKO alleles were purchased from Jackson Laboratory (Bar Harbor, ME, USA). Din-flox mice were crossbred with K14-Cre (stock #004782), Wnt1-Cre2 (stock #022501), 2.3Kb Col1a1-CreER^T2^ (stock #016241) and Gli1-CreER^T2^ (stock #007913) mice to generate K14^Cre/+^;Din^flox/flox^, Wnt1^Cre2/+^;Din^flox/flox^, 2.3KbCol1a1^CreERT2/+^;Din^flox/flox^, and Gli1^CreERT2/+^;Din^flox/flox^ cKO mice, respectively. To induce CreER^T2^ expression, tamoxifen was administrated to mice by i.p. injection (75 mg/kg) at postnatal 5 days (P5) for 3 days.
X-ray and Micro-CT
The dissected mandibles from 6-week-old mice were analyzed using X-ray radiography (Faxitron MX-20DC12, Tucson, AZ, USA). Micro-CT analyses were performed using a Scanco micro-CT35 imaging system (Scanco Medical, Bassersdorf, Switzerland) with a medium-resolution scan (7.0 μm slice increment) on mandibles. The.isq files obtained were read with KHKS-Scanco-ISQ-File Reader plugins installed in ImageJ. The.isq files were converted to.ids files using ImageJ. The.ids file obtained were converted into.ims file using Imaris File Converter. The.ims file was opened in the imaris Bitplane 9.01. Gross and sectional μ-CT imaging were obtained with pseudocolor rendering using the Imaris software.
Histology
FFPE Tissue Sections
P15 and 6 weeks-old mice were harvested and mandibles were dissected and fixed with 4% paraformaldehyde (PFA) (Sigma Aldrich Corporation, St. Louis, MO, USA) in 0.1 M phosphate buffer solution (PBS) for 48 hours and then decalcified in 10% EDTA/PBS (pH 7.4) (Sigma Aldrich Corporation, St. Louis, MO, USA) at 4°C for 2 weeks. The tissues were processed for paraffin embedding, and 5 μm sections were prepared for hematoxylin & eosin (H&E) staining and other histological analyses.
RNAscope Staining of Gli1 and Din
Dual RNAscope staining was carried out with the RNAScope 2.5 HD Brown Duplex Reagent Kit (Advanced Cell Diagnostics, 322500, Newark, CA, USA) on 5-μm FFPE tissue sections prepared from mandibles of P15 mice according to manufacturer’s instructions. Briefly, slides were baked for 1 hour at 60 °C before use. After deparaffinization and dehydration, the tissues were air-dried and treated with a hydrogen peroxidase blocker before boiling at 100–104 °C in target retrieval reagents for 15 min. Protease was then applied for 30 min at 40 °C. Target probes LacZ and Gli1 (Advanced Cell Diagnostics, 461191, 311001) were hybridized for 2 h at 40 °C, followed by a series of signal amplification and washing steps. All hybridizations at 40 °C were performed in HybEZ^™^ II Hybridization System (Advanced Cell Diagnostics). RNA staining signal was identified by two colors: green chromogenic dots developed by HRP for the Gli1 probe and red chromogenic dots developed by AP for the LacZ probe. Following the RNAscope assay, samples were counterstained for 2 min with hematoxylin.
Cryosection and X-galactosidase (Gal) Staining
The Din-KO allele has a LacZ reporter driven by endogenous Din promoter, which can be used for indicating Din expression (Fig. 1A. tm1b). X-Gal Staining was performed on the cryosections of mandibles obtained from 4 weeks old Din-KO heterozygous (normal) mice as described previously^39, 46^. Briefly, tissues for cryosection were fixed in 4% PFA for 24 hours at 4 °C followed by decalcification in 10% EDTA for 2 weeks. 30% sucrose dehydration at 4 °C overnight followed by OCT embedding for cryosection. Cryo-sections were fixed with 0.2% glutaraldehyde in PBS at 4 °C for 30 min. The sections were washed for three times in 0.005% NP-40 and 0.01% sodium deoxycholate and then incubated in staining solution (5 mM potassium ferrocyanide and potassium ferricyanide, 2 mM MgCl2, 0.4% X-Gal in dimethylformamide) at 37 °C for 3–24 hours. Post fixation was done with 4% PFA in PBS at room temperature for 1 hour followed by counterstaining with nuclear fast red (Vector Laboratories Inc, Newark, CA, USA)
Cell Proliferation (EdU) and TUNEL Assays
Mice were i.p. injected with EdU (50 mg/kg in PBS, ThermoFisher, C10352, Carlsbad, CA, USA). The mandibles were collected 2 h after injection and processed for cryosection or FFPE section. EdU incorporation was detected using a Click-iT Kit (ThermoFisher Scientific, C10337, Waltham, MA, USA) as we previously described^39^. For TACs assay, mice were injected EdU at P10, P15 and P21 days.
Apoptotic cells were examined on the FFPE sections via TUNEL staining using an ApopTag Plus In Situ Apoptosis Fluorescein Detection Kit (Millipore, S7111, Burlington, MA, USA) according to the manufacturer’s instruction. DAPI was used as counterstaining.
Immunofluorescece Staining of DSPP and AMEL with EdU
EdU injection, mandible collection, paraffin embedding, FFPE section and IF staining were performed as described above. IF staining of DSPP and AMEL was performed as previously described^40^. The primary antibodies were anti-DSPP (1:200, Abcam, ab216892, Burlingame, CA, USA) and anti-AMEL (1:500, Santa Cruz Biotechnology, sc-32892, Dallas, TX, USA). Primary antibodies were detected using AlexaFluor488 conjugated secondary antibody and AlexaFluor633 conjugated secondary antibody (ThermoFisher, A-11034, and A-21070). DAPI was used for counterstaining.
Label Retaining Cell Assay
Din-KO and WT mice were i.p. injected EdU (50 mg/kg) at P10 for consecutive 7 days, and sacrificed at postnatal 8 weeks as described previously^47^. Briefly, the mandibles were fixed with 4% PFA followed by decalcification and PEGASOS tissue clearing^48^. Fluorescent imaging was acquired by an SP8 Confocal Microscope (Leica Microsystems, Wetzlar, Germany). Image processing and 3D rendering were performed with Imaris 9.0 (Bitplane, Belfast, UK) as described previously^47^.
Single-Cell RNA Sequencing (scRNA-seq)
Preparation of Single Cell Suspension
The proximal tissues of the lower incisors of P15 Din-KO mice and WT mice were obtained with fine forceps in ice-cold PBS. The tissue was minced into 0.5 mm pieces and dissociated into single-cell suspension by digesting in a-MEM containing 2.5 mg/ml liberase and 1U/μl DNase at 37C° for 30 min, then terminated with 10% FBS and passed through Pluri-strainer Mini 70μm (PluriSelect, 43-10070-40, El Cajon, CA, USA). Dead cells were removed using the Dead Cell Removal Kit (Miltenyi Biotec, 130-090-101, MD, USA).
Library Construction and Sequencing
Cell barcoding, cDNA synthesis, and library preparation were done following the protocols of 10X Next GEM single-cell 3’ reagent kits V3.1 (10× Genomics, 1000128, Peasanton, CA, USA). Quality control for cDNA and library was performed on a 2100 Bioanalyzer (Agilent Technology). Sequencing was performed with NovaSeq PE-150 at LC Sciences (Houston, TX, USA).
Bioinformatics Analysis of scRNAseq Data
Sequencing results were demultiplexed and converted to FASTQ format using Illumina bcl2fastq software. The CellRanger (https://support.10xgenomics.com/single-cell-gene-expression/software/pipelines/latest/what-is-cell-ranger) was used to perform barcode processing and 3’ gene counting. The cDNA insert was aligned to the mm10/GRCm38 reference genome. Only confidently mapped, non-PCR duplicates with valid barcodes and UMIs were used to generate the gene-barcode matrix. Further analysis included quality filtering, the identification of highly variable genes, dimensionality reduction, and standard unsupervised clustering. Low-quality cells with fewer than 1,000 detected genes were removed. Cells with more than 10% of the transcripts coming from mitochondrial genes or more than 3% Hbb/Hba content were also removed. Dimensionality reduction was performed with UMAP as described previously^49^. A joint analysis of Din-KO and WT data was performed following the Seurat integration procedure^50, 51^. Cell-cycle scoring and regression were performed with the default list of cell-cycle genes. Clustering was performed with ‘FindClusters’ and ‘resolution=1’. Cluster marker genes were identified with ‘FindAllMarkers’. Trajectory analyses were carried out on the odontogenic subset using Monocle 3 and TradeSeq^52, 53^. Cell-cell communication analysis were performed using CellChatand CellphoneDB v2.1.4^54, 55^.
Re-visiting Open-Access scRNA-seq Data
The raw scRNA-seq data matrices of dental cell type atlas available online from PK lab, Harvard University, were analyzed through bioinformatics using the R codes for retrieving each cluster from the data matrix to examine the expression pattern of Din in different cell populations in mouse incisors. Dimensionality reduction and cluster annotation information of cell subgroups in the whole incisor was performed and the cell subgroups expressing Din were analyzed tSNE embedding and cluster annotation information was obtained at: http://pklab.med.harvard.edu/ruslan/dental.atlas.html. R codes for retrieving each cluster from the matrix were obtained at: http://pklab.med.harvard.edu/ruslan/tooth_atlas/scripts/. The data analysis protocol was followed as described previously^56^.
Lineage Tracing of Gli1+ MSCs
Gli1-CreER^T2^ and td-Tomato mice were crossbred with Din-KO mice to generate Gli1^CrERT2/+^;Din-KO^+/−^;td-Tomato (heterozygous normal) and Gli1^CrERT2/+^;Din-KO^−/−^;td-Tomato (homozygous KO) mice. CreER expressions were induced by a single injection of tamoxifen (75 mg/kg, i.p.) at P11, and td-Tomato expressions were traced for 36 h, 3 days and 7 days. The tissues were harvested and processed for cryosections. 10 μm cryosections were prepared in cryotome (Leica, CM 1860) and processed for fluorescent microscopy using a scanning microscope (Olympus, BX61VS Slide Scanners, Center Valley, PA, USA).
Incisor Injury Repair Assay
6-weeks old Din-KO mice and their heterozygous (normal) littermates were used for injury repair assays. The mice were anesthetized with isoflurane. The left lower incisors were clipped at the level of gingival papilla by removing the erupted part. The length of broken incisors was measured with a digital caliper (VWR, Radnor, PA, USA) on daily basis for 10 days.
Primary Culture of Dental Pulp Gli1+ MSCs and CFU-F Assay
Incisor pulp was obtained from 6 weeks-old Gli1^CrERT2/+^;Din-KO^+/−^;td-Tomato (heterozygous normal) and Gli1^CrERT2/+^;Din-KO^−/−^;td-Tomato (homozygous KO) mice after tamoxifen induction for 2 days. The dental epithelium was carefully removed with fine forceps.The pulp tissue was minced into 0.5 mm pieces and dissociated into single-cell suspension by digesting in a-MEM containing 2.5 mg/ml liberase and 1U/μl DNase at 37C° for 30 min, then terminated with 10% FBS and passed through Pluri-strainer Mini 70μm (PluriSelect). The cell suspension was sorted using the FACSAria II (BD Biosciences, Franklin Lakes, NJ, United States).
For CFU-F (colony forming unit-fibroblasts) assay, the Gli1+ MSCs obtained from cell sorting were seeded in a 100 mm plate at approximately 1million cells per 10 ml of growth medium. The cells were grown for 3 days and the growth medium changed to remove unattached cells and selectively grow the adherent MSCs. The adherent MSCs were further grown until the 5^th^ day to reach approximately 70% confluency. Passage 2 MSCs were seeded in 6 well plates at a low density of 1×10^4^ cells and grown in growth media containing a-MEM, 10% FBS, 1% antibiotic-antimycotic for 3 weeks. Cells were washed with PBS and fixed with 100% methanol. Cells were stained with crystal violet solution. Gross photographs of the stained plates were taken and the CFU-F colonies were counted manually.
Tri-lineage Differentiation Assay
For osteogenesis assay, the Gli1+ MSCs obtained from cell sorting were cultured in 6 well plates at a density of 4.2 ×10^3^ cells/cm^2^ in 2 ml StemXVivo Osteogenic/Adipogenic Base Media (Bio-Techne, CCM007, Minneapolis, MN, USA) with 1% antibiotic-antimycotic (ThermoFisher) until 70 % confluency was reached. The cells were further grown in differentiation media with StemXVivo Mouse/Rat Osteogenic supplement (Bio-Techne, CCM009) for 3 weeks with media changed every 3 days. The cells were stained with Alizarin Red S and the photograph was obtained. The cells were scrapped with 10 % acetic acid, mixed homogenously in a vortex, and centrifuged, the colored supernatant was collected and neutralized with 10% ammonia water. The colorimetric assay was performed to obtain the OD values using a Biotek Cytagen Gen5 microplate reader (Agilent, Santa Clara, CA, USA) and the differences were shown in statistics.
For adipogenesis assay, the MSCs in StemXVivo Osteogenic/Adipogenic Base Media reached 100 % confluency were further grown in differentiation media made with Human/Mouse/Rat StemXVivo^®^ Adipogenic Supplement (Bio-Techne, CCM011) for one week with media changed every 3 days. The cells were stained with Sudan Red III and counterstained with Mayers Hematoxylin. The bright field imaging of cells was done under the inverted microscope. Random pictures of cells were taken from 10 different microscopic fields and the number of red-stained adipocytes were counted manually between Din-KO and WT cells.
For chondrogenesis assay, 1.25 × 10^5^ MSCs in StemXVivo Chondrogenic Base Media (Bio-Techne, CCM005) were centrifuged at 200 × g for 5 min to pellet the cells. The pellets in tubes were incubated upright in completed StemXVivo Chondrogenic Differentiation Media (Bio-Techne, CCM006) at 37° C and 5% CO_2_ for 2 days with media changed every 2–3 days. Chondrogenic pellets were harvested after 20 days in culture and imaged under the inverted microcope.
Cell Scratch Motility Assay
Din-KO and WT MSCs were cultured in 6 well plates in StemXVivo Mesenchymal Stem Cell Expansion Media (Bio-Techne, CCM004) to obtain a cell monolayer of 70 % confluency. Plastic pipette tip was used to make a straight scratch line on cells monolayer. Cell migration towards the scratch line were observed in 0 , 5 and 10 h and microscopic photographs taken. The distance between two scratch lines in the photographs were measured in Image J software to determine differences in the cell migration distance between Din-KO and WT MSCs.
Senescence β-Gal Staining
Din-KO and WT MSCs were cultured in 12 well plates in StemXVivo Mesenchymal Stem Cell Expansion Media to obtain a cell monolayer of 80 % confluency. The senescence of Din-KO and WT MSCs was examined by β-Gal Staining kit (Cell Signaling, 9860, Cell Signaling, Danvers, MA, USA) following the manufacturer’s instructions. Random pictures of cells were taken from 10 different microscopic fields and the number of blue-stained senescence cells were counted manually between Din-KO and WT cells.
Q-PCR
The proximal tissues of lower incisors were dissected from postnatal 0, 7, 14, and 28-day-old Din-KO and WT mice using fine forceps under a stereo microscope. The tissues were flash-frozen in liquid nitrogen, crushed using a mortar and pestle, and total mRNA was extracted using a RNeasy Kit (QIAGEN, 74104, Germantown, MD, USA) following the manufacturer’s protocol. 1 μg total RNA was used to make cDNA using a Reverse Tanscriptase kit (QIAGEN, 220211). P16 and P21 qPCR primers were designed, and the mRNA levels were determined using quantitative real-time PCR (qRT-PCR) analysis performed in triplicates using the SYBR Green method in the Biorad CFX 100 qPCR analyzer. The housekeeping gene, gapdh, was used to normalize the expression levels of target genes.
Microscopy
Bright-field images were acquired by an Olympus CX43 Upright Light Microscope or an Olympus CKX41 inverted microscope connected with a DP27 imaging system (Olympus). Immunofluorescent images were acquired by an Olympus BX61VS Slide Scanner (Olympus) or an SP8 confocal microscope (Leica).
Mammalian Expression Vectors
pCMV6-Din-GFP vector:
The cDNA of mouse Din (4930453N24Rik) was synthesized by GenScript with HindIII and MluI restriction sites on 5’ and 3’ ends and subcloned into pCMV6-AC-GFP vector (OriGene, PS100010, Rockville, MD, USA) to form a pCMV6-Din-GFP vector for mammalian expression.
pCMV6-Din-HA Vector:
The GFP sequence in pCMV6-Din-GFP vector constructed above was removed by MluI and PmeI restriction enzymes. An HA cDNA sequence synthesized by GenScript with MluI and PmeI restriction sites on 5’ and 3’ was subcloned into the GFP-removed notch to form a pCMV6-Din-HA vector for mammalian expression.
Transient and Stable Cell Transfection
For transient expression of Din-GFP fusion proteins, HEK293 and NIH/3T3 cells were seeded in a 12-well plates in 1×10^5^/cm^2^ density, and grown for 70% confluency in a growth medium containing α-MEM, 10% Heat Inactivated FBS and 1% antimycotic-antibiotic. The pCMV6-Din-GFP plasmid was transfected to the cells using Lipofectamine 3000 (ThermoFisher, L3000001) in Opti-MEM media (ThermoFisher, 31985070) following the manufacturer’s instructions. The cells were fixed with 10% neutral buffered formalin and permeabilized with 0.1% Triton-X 100 in PBS and immunostained with β-Actin primary antibody (Cell Signaling, 4967) and nuclei counterstained with Gold Antifade Reagent with DNA Stain DAPI (ThermoFisher, P36931). Imaging was done using an SP8 confocal microscope (Leica).
For stable expression of Din-HA fusion proteins, NIH/3T3 cells transfected with pCMV6-Din-HA plasmids were selected with neomysin following ThermoFisher Stable Transfection Protocol. The expression of DIN-HA fusion proteins was confirmed by Western Blotting using anti-HA antibody (ThermoFisher, 26183). Cells stably transfected with pCMV6-AC-GFP plasmids were used as blank control.
Co-Immunoprecipitation and Mass Spectrometry
Cells stably expressing Din-HA were harvested at 90% confluency and lysed on ice for 15 min. Co-IP was performed using Pierce^™^ HA-Tag IP/Co-IP Kit (ThermoFisher, 26180) according to the manufacturer’s instructions. The precipitated proteins with DIN-HA were eluted with SDS sample buffer and heated at 95C° for 10 min and subjected to BCA quantitation. Equal amounts of proteins from each group were loaded on SDS-PAGE. Coomassie Blue stained protein bands were cut off and diced into ~1 mm^3^ pieces, then digested overnight with trypsin followed by reduction and alkylation with DTT and iodoacetamide. After solid-phase extraction clean-up, 2 μl of each sample was examined on a QExactive HF mass spectrometer coupled to an Ultimate 3000 RSLCNano liquid chromatography system (LC-MS) (ThermoFisher).
The LC-MS data were analyzed with Proteome Discoverer v2.4 (ThermoFisher), with peptide identification performed using Sequest HT searching against the mouse protein database from UniProt. Fragment and precursor tolerances of 10 ppm and 0.02 Da were specified, and three missed cleavages were allowed. Carbamidomethylation of Cys was set as a fixed modification, with oxidation of Met set as a variable modification. The false-discovery rate cut-off was 1% for all peptides. Proteins that were identified with only 1 peptide-spectrum match, identified but not quantified, or with potential contaminants were excluded from further analysis. The downstream analysis followed the workflow of the DEP package^57^. Protein-wise linear models combined with empirical Bayes statistics were used for the differential expression analysis. Empty vector IPs were used as experimental controls to provide a background list of proteins binding non-specifically to the construct. Proteins were filtered for protein fold change equal to or greater than 2.0. GO enrichment of differentially expressed proteins was performed using the Clusterprofile R package.
G-LISA Assays
The dental pulpMSCscells isolated above were subjected to serum starvation for 48 h to lower the basal levels of GTPase activity and increase the response to a given activator. The cells were then activated with a 0% FBS growth medium containing Rho/Rac/CDC42 Activator I (Cytoskeleton Inc., CN04-A, Denver, CO, USA) for 2.5 hours, and with EGF for 3 min (Cytoskeleton, CN02) for Ras GTPases activation. The cells without RhoA/Rac/CDC42 Activator I and EGF were used as a control to measure the basal GTPase levels. Immediately the cells were washed with 10 ml ice-cold Ca^+^ and Mg^++^ free DPBS and lysed in 600 μl ice-cold cell lysis buffer on ice following the manufacturer’s instructions. The cells were scraped and collected in prechilled 1.5ml EP tubes and centrifuged at 10,500 rpm at 4°C for 1 min. The supernatant obtained was immediately aliquoted in multiple prechilled 1.5 ml EP tubes on ice. The cell lysates aliquots were flash-frozen in liquid nitrogen and stored at −80°C. The protein concentration of each sample was measured as per the manufacturer’s instructions using the 600 nm wavelength of BioTek Cytation 5 Spectrophotometer (Agilent). Ras/RhoA/Rac1/CDC42 GLISA Assay was performed adhering to the manufacturer’s instructions and recommendations. Briefly, each protein sample concentration was equalized using the respective cell lysis buffer and subjected to Ras/RhoA/Rac1/CDC42 GLISA Assay. The final Ras/RhoA/Rac1/CDC42 activity was measured using the 490 nm wavelength of BioTek Cytation 5 Spectrophotometer comparing WT and Din-KO MSCs.
Protein Domain Prediction
Phyre2 and cNLS Mapper were employed for protein domain prediction.
Statistics
At least three independent samples were used in analysis wherever required and the data were expressed as mean ± SD. Unpaired Student’s t-test and one-way ANOVA were used to analyze data sets with two groups and when comparing more than two groups respectively. For one-way ANOVA, p-values were determined followed by Tukey’s multiple comparison tests. Prism software package (GraphPad Prism 9) or Microsoft Excel were used in statistical calculations. *P<0.05, **P<0.01, ***P< 0.001, and ****P<0.0001 were used to determine statistically significant differences.
Supplementary Files
This is a list of supplementary files associated with this preprint. Click to download.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Sweet H.O., Marks S.C., Mac Kay C.A., Johnson K.R., Davisson M.T. Dense incisors (din): a new mouse mutation on chromosome 16 affecting tooth eruption and body size. J. Hered. 87, 162–167 (1996).8830097 10.1093/oxfordjournals.jhered.a 022977 · doi ↗ · pubmed ↗
- 2Fairfield H. Exome sequencing reveals pathogenic mutations in 91 strains of mice with Mendelian disorders. Genome. Res. 25, 948–957 (2015).25917818 10.1101/gr.186882.114PMC 4484392 · doi ↗ · pubmed ↗
- 3Shi C. BMP Signaling in Regulating Mesenchymal Stem Cells in Incisor Homeostasis. J. Dent. Res. 98, 904–911 (2019).31136721 10.1177/0022034519850812 PMC 6616121 · doi ↗ · pubmed ↗
- 4Du J. Arid 1a regulates cell cycle exit of transit-amplifying cells by inhibiting the Aurka-Cdk 1 axis in mouse incisor. Development 148, dev 198838 (2021).33766930 10.1242/dev.198838 PMC 8077510 · doi ↗ · pubmed ↗
- 5Jing J. Reciprocal interaction between mesenchymal stem cells and transit amplifying cells regulates tissue homeostasis. Elife 59459 (2021).10.7554/e Life.59459 PMC 782259333480845 · doi ↗ · pubmed ↗
- 6Yang G. Mesenchymal TGF-β Signaling Orchestrates Dental Epithelial Stem Cell Homeostasis Through Wnt Signaling. Stem Cells 32, 2939–2948 (2014).24964772 10.1002/stem.1772 · doi ↗ · pubmed ↗
- 7Chen S. Runx 2+ Niche Cells Maintain Incisor Mesenchymal Tissue Homeostasis through IGF Signaling. Cell Rep. 32, 108007 (2020).32783935 10.1016/j.celrep.2020.108007 PMC 7461627 · doi ↗ · pubmed ↗
- 8Smith C.E., Warshawsky H. Cellular renewal in the enamel organ and the odontoblast layer of the rat incisor as followed by radioautography using 3H-thymidine. Anat. Rec. 183, 523–561 (1975).1200409 10.1002/ar.1091830405 · doi ↗ · pubmed ↗