RNA processing kinase inhibitors and epigenetic inhibitors in combination with oncology drugs or investigational agents in multi-cell type patient-derived tumor cell line spheroids
Beverly A. Teicher, Thomas S. Dexheimer, Thomas Silvers, Nathan P. Coussens, Eric Jones, Steven D. Gore, Mark Kunkel, James H. Doroshow

TL;DR
This study explores how combining RNA processing and epigenetic inhibitors with cancer drugs affects tumor cell spheroids, showing enhanced cytotoxic effects.
Contribution
The novel contribution is the demonstration of additive or greater-than-additive effects of CLK and LSD1 inhibitors with anticancer drugs in multi-cell tumor spheroids.
Findings
CLK inhibitors combined with anticancer drugs showed enhanced cytotoxicity in tumor spheroids.
LSD1 inhibition was effective when combined with ubiquitin proteasome pathway inhibitors.
The KRAS G12D inhibitor MRTX-1133 showed activity in tumor lines with the KRAS G12D mutation.
Abstract
The alternative splicing of mRNA precursors allows one gene to yield multiple proteins with distinct functions. CDC-like kinases (CLKs) serve as pivotal regulators of alternative splicing. Control of protein expression also occurs at the level of DNA through histone methylation and demethylation. We investigated the activity of two CLK inhibitors, cirtuvivint and CC-671, and the LSD1 inhibitor iadademstat alone and in combination with anticancer drugs or investigational agents. Well-characterized patient-derived cancer cell lines from the PDMR (https://pdmr.cancer.gov/models/database.htm) were used along with standard human cancer cell lines. Multi-cell type (mct) tumor spheroids were grown from a ratio of 6:2.5:1.5 malignant cells, endothelial cells, and mesenchymal stem cells. Following three days of growth, the spheroids were exposed to the single agents or combinations at…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
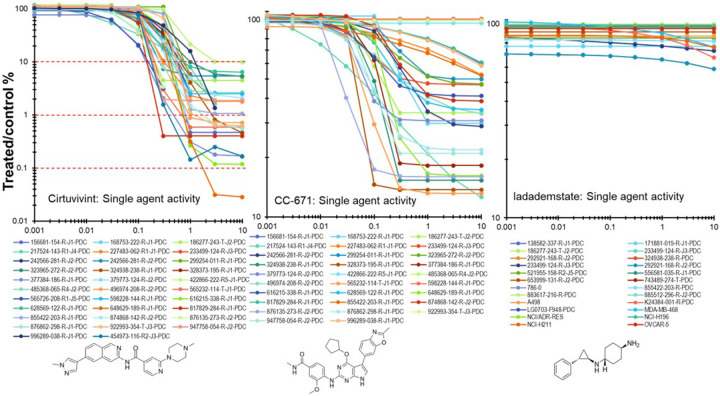 Figure 1
Figure 1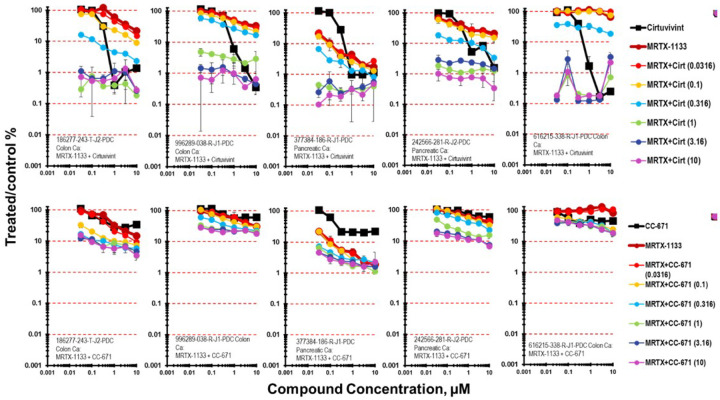 Figure 2
Figure 2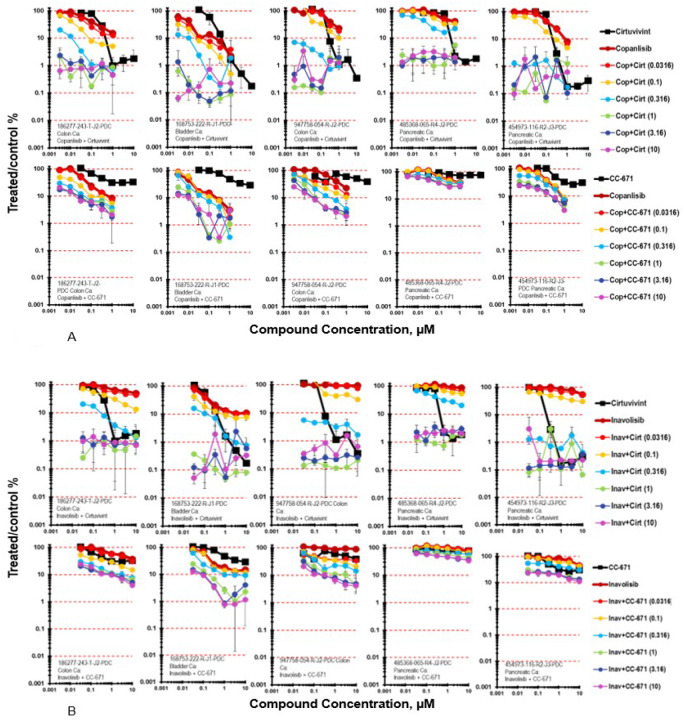 Figure 3
Figure 3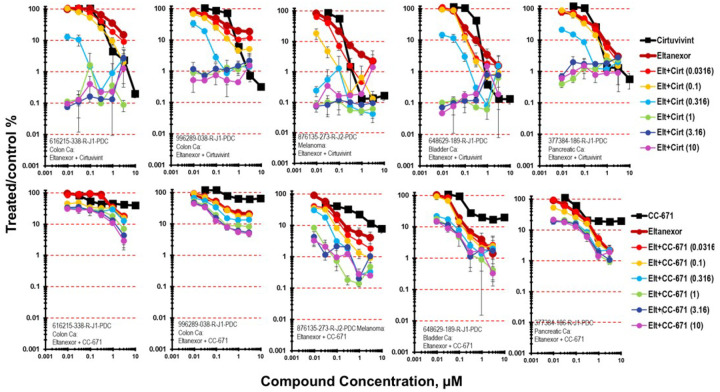 Figure 4
Figure 4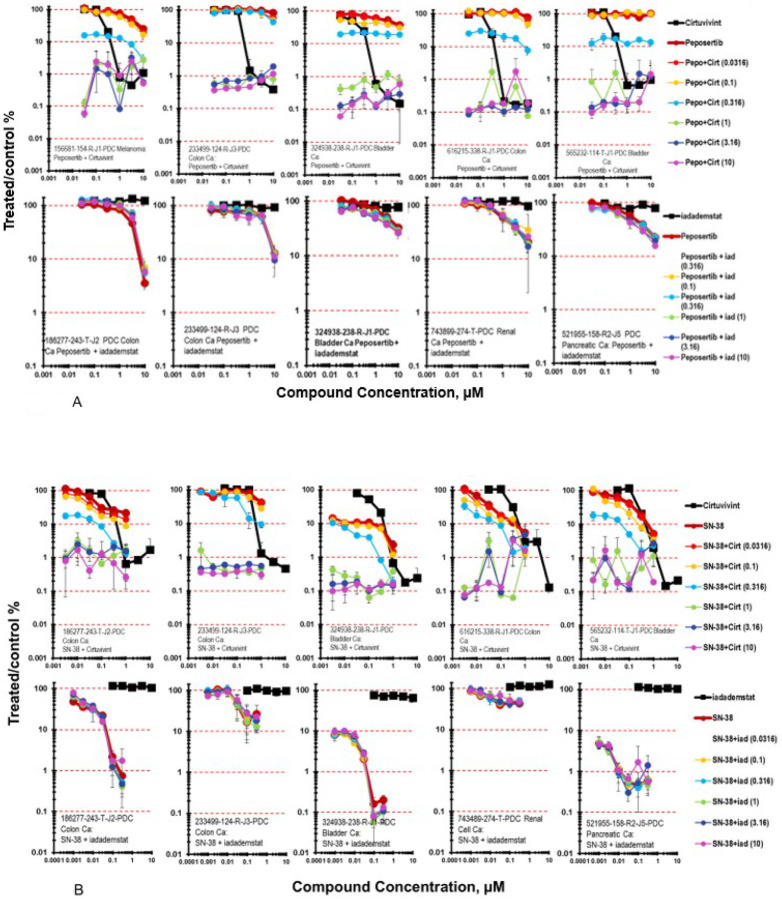 Figure 5
Figure 5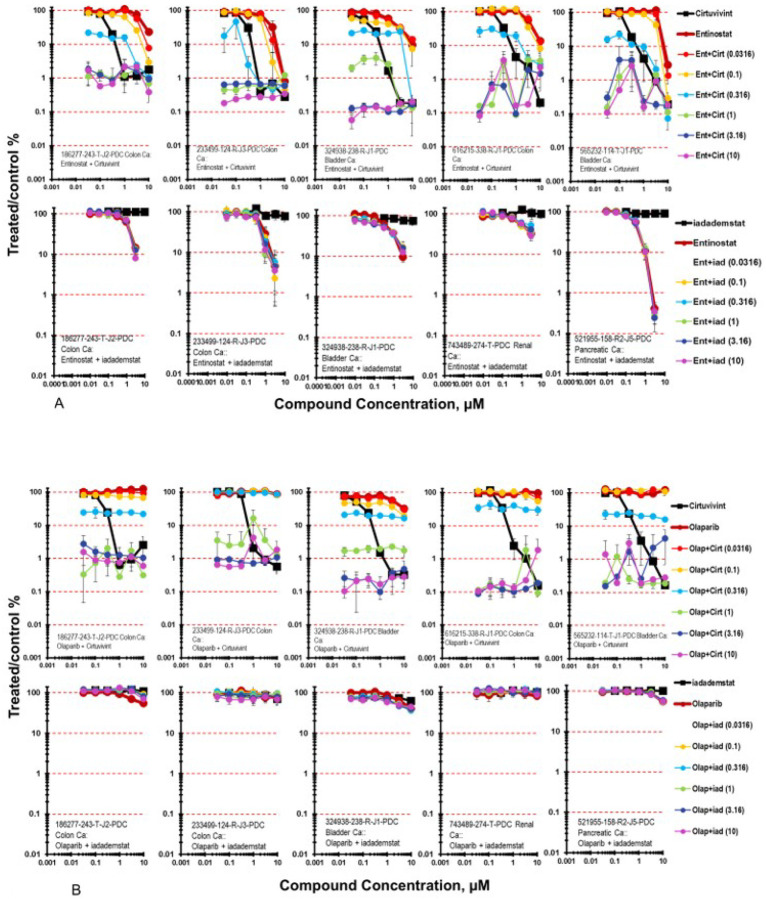 Figure 6
Figure 6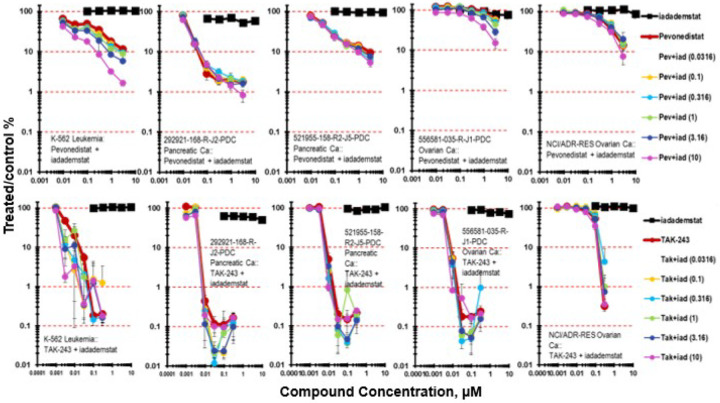 Figure 7
Figure 7Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsUbiquitin and proteasome pathways · Protein Degradation and Inhibitors · Histone Deacetylase Inhibitors Research
INTRODUCTION
The spliceosome, a critical intracellular organelle, is multi-megadalton complex composed of over 100 proteins including 5 small nuclear ribonucleoproteins. Cancer cells often have cancer type-specific RNA splicing alterations. Most multi-exon human genes undergo alternative splicing, allowing multiple mature mRNAs to be derived from a single gene [1, 2]. Thus, 25,000 genes can code for well over 100,000 proteins. Pre-mRNA splicing, the process of removing introns from precursor messenger RNA, is critical in the post-transcriptional regulation of gene expression [3]. Serine/arginine (SR) family proteins control the patterns of alternative splicing in pre-mRNA and enhance splicing from nearby splice sites by interacting with exonic and intronic splicing enhancer sequences in pre-mRNA [3]. SR-proteins require phosphorylation by SR protein kinases (SRPKs), protein kinase B (PKB/AKT), NIMA-related kinase2 (NEK2), PRP4 kinase (PRP4K) dual-specificity tyrosine phosphorylation-regulated kinase 1A (DYRK1A), cAMP-dependent protein kinase (PKA) or by the CDC-like kinases (CLKs) to be active [4]. The CLKs regulate transcript RNA splicing through SR protein phosphorylation [5]. Cirtuvivint is a pan CDC-like kinase (CLK1-4) and dual specificity tyrosine kinase (DYRK1-4) inhibitor which targets mRNA splicing and the Wnt pathway which is in clinical trial [6]. Cirtuvivint exposure disrupts spliceosome activity and decreases production of Wnt signal pathway splicing variants. A Phase I clinical trial of cirtuvivint by oral administration, assessing dose escalation is currently active but not recruiting (NCT03355066) and the combination of cirtuvivint with ASTX727 in patients with acute myeloid leukemia or myelodysplastic syndromes is actively accruing patients (NCT06484062) [5]. CC-671, a CLK/TTK inhibitor has an inhibitory repertoire similar to cirtuvivint. TTK, also called Monopolar spindle 1 (Mps1), is a dual serine/threonine kinase that regulates the spindle assembly checkpoint, controlling cellular progression through mitosis [7, 8]. CC-671 had in vivo activity in human tumor xenografts due to dual inhibition of CLK2/TTK [4].
Lysine-specific demethylase 1α (LSD1) encoded by the KDM1A gene, is a lysine demethylase which has recurrent mutations, translocations, and somatic copy number gains or losses in human tumors [9, 10]. LSD1/KDM1A removes mono- and dimethyl groups of histone H3 lysine-4 (H3K4), lysine-9 (H3K9) as well as non-histone substrates [11, 12]. LSD1 interacts with other chromatin regulators including histone deacetylases (HDAC)-1, 2 and 3, and DNA methyltransferase 1 (DNMT1). LSD1 is a component of a multi-subunit complex causing transcription activation or repression [13]. LSD1 non-histone substrates are associated with the regulation of cell cycle progression and apoptosis. LSD1/KDM1A expression is associated with poor prognosis in prostate, breast, lung, bladder, colorectal cancer and neuroblastoma. Iadademstat is a potent LSD1 inhibitor with an IC_50_ of 18 nM and > 1000-fold higher selectivity towards LSD1 compared to related FAD-dependent aminoxidases [14, 15]. In AML models, LSD1 inhibition did not alter genome methylation but did increase me_2_H3K4 at LSD1 target genes [16–18]. In a small cell lung cancer xenograft, treatment with iadademstat resulted in NOTCH activation and ASCL1 suppression decreasing neuroendocrine properties [19].
With the complexity of cancer genomic alterations, single targeted drugs are usually not sufficient to impact malignant disease, and combinations of targeted drugs are necessary for effective treatment. The current study was undertaken to explore the activity of RNA processing and epigenetic inhibitors in combination with other targeted agents and standard-of-care drugs in a mct-spheroid model from patient-derived tumor cell lines of the PDMR (https://pdmr.cancer.gov/) or standard tumor cell lines and stromal cells. The full data sets are available at the PubChem files listed on Table 4.
MATERIALS AND METHODS
Compounds.
The drugs and investigational agents: cirtuvivint (SM08502; NSC835563), CC-671 (NSC850746), iadademstat (NSC806812), vemurafenib (NSC761431), tapotoclax (NSC804041), sotorasib (NSC818433), adagrasib (NSC831453), MRTX-1133 (NSC836407), BAY2416964 (NSC825713), copanlisib (NSC816437), inavolisib (NSC800729), abemaciclib (NSC768073), osimertinib (NSC779217), entinostat (NSC756642), eltanexor (KPT-8602, NSC794443), venetoclax (NSC766270), ceralasertib (NSC777638), bortezomib (NSC756655), olaparib (NSC753686), talazoparib (NSC767125), adavosertib (NSC754677), alisertib (NSC759677), ART-558 (NSC835418), aza-T-dCyd (NSC777586), AZD-1390 (NSC803789), belinostat (NSC758774), camonsertib (NSC841442), CPI-455 (NSC825282), decitabine (NSC127716), elimusertib (NSC800525), ixazomib (NSC758254), KSQ-4279 (NSC840948), panobinostat (NSC761190), peposertib (NSC802822), pevonedistat (NSC761192), R306465 (NSC773264), selinexor (NSC780203), TAK-243 (NSC785004), tazemetostat (NSC777109), topotecan (NSC609699), TP-3654 (NSC805149), eribulin (NSC707389), etoposide (NSC141540), carboplatin (NSC241240), cisplatin (NSC119875), gemcitabine (NSC613327), 5-fluorouracil (NSC19893), SN-38 (NSC673596), oxaliplatin (NSC266046), paclitaxel (NSC125973), and doxorubicin (NSC123127), were obtained from the National Cancer Institute (NCI) Developmental Therapeutics Program Chemical Repository [20]. The FDA-approved anticancer drug set is available from the Developmental Therapeutics Program at https://dtp.cancer.gov/organization/dscb/obtaining/available plates.htm. The drugs and investigational agents used were demonstrated to be >95% pure by proton nuclear magnetic resonance and liquid chromatography/mass spectrometry. The stock solutions were prepared in dimethyl sulfoxide (DMSO, Sigma-Aldrich, St. Louis, MO, cat. D2650), except for the platinum complexes which were prepared in saline (Quality Biological, Gaithersburg, MD, cat. 114-055-101), at 800-fold the tested concentration and stored at −70°C prior to their use. All drugs and investigational agents were tested over a range starting from a high concentration at or near the clinical C_max_ and decreasing in half-log increments. If the clinical C_max_ for an agent had not been determined, the highest concentration tested was 10 μM (Table 1).
Cell Lines.
The patient-derived cancer (PDC) cell lines include 16 colon adenocarcinoma lines: 186277-243-T-J2-PDC, 188146-221-R-J1-PDC, 282377-053-R-J1-PDC, 381356-305-R-J1-PDC, 435261-313-R-J1-PDC, 439559-082-T-J2-PDC, 519858-162-T-J1-PDC, 616215-338-R-J1-PDC, 695427-040-R-J1-PDC, 233499-124-R-J2-PDC, 817829-284-R-J1-PDC, 825966-067-R-J1-PDC, 857933-349-R-J2-PDC, 996289-038-R-J1-PDC, 997537-175-T-J1-PDC, and 947758-054-R-J2-PDC; 16 pancreatic carcinoma lines: 217524-143-R1-J4-PDC, 227483-062-R1-J1-PDC, 242566-281-R-J2-PDC, 292921-168-R-J2-PDC, 323965-272-R-J2-PDC, 377384-186-R-J1-PDC, 422866-222-R5-J1-PDC, 454973-116-R2-J3-PDC, 485176-168-R4-J1-PDC, 485368-065-R4-J2-PDC, 521955-158-R2-J5-PDC, 777334-354-R1-J3-PDC, 885724-159-R-J1-PDC, 966289-007-R4-J1-PDC, K24384-001-R-PDC, and 496974-208-R-J2-PDC; 6 bladder carcinoma lines: 168753-222-R-J1-PDC, 324938-238-R-J1-PDC, 565232-114-T-J1-PDC, 648629-189-R-J1-PDC, 883617-216-R-PDC, and 855422-203-R-J1-PDC; 7 endometrial carcinomas: 379773-124-R-J2-PDC, 598228-144-R-J1-PDC, 633275-114-R-J1-PDC, 636577-100-R-J1-PDC, 922993-354-T-J2-PDC, and 876862-298-R-J1-PDC; 3 head & neck squamous carcinoma: 328373-195-R-J1-PDC, 628569-122-R-J1-PDC, and 874868-142-R-J2-PDC; 4 melanoma: 156681-154-R-J1-PDC, 276233-004-R-J1-PDC, 299254-011-R-J1-PDC, and 876135-273-R-J2-PDC; 3 NSCLC: 349418-098-R-PDC, 653999-131-R-J2-PDC, and LG0703-F948-PDC; 2 breast carcinoma: 171881-019-R-J1-PDC and 885512-296-R-J2-PDC; 2 MPNST: 317291-083-R-J1-PDC and 596521-263-R-J1-PDC; 1 ovarian carcinoma: 556581-035-R-J1-PDC; 1 renal cell carcinoma: 743489-274-T-PDC; and 1 Merkel cell tumor: 138582-337-R-J1-PDC, were obtained from the NCI Patient-Derived Models Repository (PDMR, https://pdmr.cancer.gov/) (Table 2). In addition, several standard human tumor cell lines were used including: 786-0 and A498 renal cell carcinoma, IGROV1, NCI/ADR-RES and OVCAR-5 ovarian carcinoma; MDA-MB-231, MDA-MB-468, and SUM149PT TNBC; NCI-H1876, NCI-H196, NCI-H211 and SW 1271 SCLC; A375 melanoma and K562 leukemia were obtained from the NCI Tumor Repository (Table 2). Pooled donor human umbilical vein endothelial cells (HUVEC, Lonza, cat. CC-2519) and human mesenchymal stem cells (hMSC, Lonza, cat. PT-2501) were purchased from Lonza (Walkersville, MD).
Cell Culture.
All cells were maintained in an incubator at 37°C and 5% CO_2_ with 95% humidity. The PDC lines were cultured according to standard operating procedures established by the NCI PDMR (https://pdmr.cancer.gov). Briefly, all PDCs were thawed and cultured in Matrigel-coated flasks prepared with a working solution of 1X Ham’s F-12 nutrient mix, without supplementation (Invitrogen, Waltham, MA, cat. 11765054), 100 U/mL penicillin-streptomycin (Invitrogen, cat. 15140122), and 2.5% Matrigel (Corning Inc., Corning, NY, cat. 354248) for the first three passages. All PDCs were cultured in complete DMEM/F-12 media containing advanced DMEM/F-12 (Invitrogen, cat. 12634028), 4.9% defined fetal bovine serum, heat inactivated (HyClone Laboratories Inc., Logan, UT, cat. SH30070.03HI), 389 ng/mL hydrocortisone (Sigma-Aldrich, cat. H4001), 9.7 ng/mL human EGF recombinant protein (Invitrogen, cat. PHG0313), 23.4 μg/mL adenine (Sigma-Aldrich, cat. A2786), 97.3 U/mL penicillin-streptomycin (Invitrogen, cat. 15140122), 1.9 mM L-glutamine (Invitrogen, cat. 25030081), and 9.7 μM Y-27632 dihydrochloride (Tocris Bioscience, Bristol, United Kingdom, cat. 1254). The PDCs were cultured in complete DMEM/F12 media without 10 μM Y-27632 dihydrochloride for at least two passages prior to the screen, unless specified otherwise (Table 3). The established cell lines were cultured in RPMI-1640 medium, HEPES (Invitrogen, cat. 22400105) with 10% defined fetal bovine serum (HyClone Laboratories Inc., cat. SH30070.03) and 2 mM L-glutamine (Invitrogen, cat. 25030081), unless specified otherwise (Table 3). The pooled donor HUVEC and hMSC were cultured in endothelial cell growth medium 2 (PromoCell, Heidelberg, Germany, cat. C-22011) and mesenchymal stem cell growth medium 2 (PromoCell, cat. C-28009). For all experiments, HUVEC and hMSCs were used at passages ≤5, while the malignant cell lines were used at passages ≤15. Samples of the cell lines were collected at regular intervals throughout the screening process for short tandem repeat (STR) profiling and mycoplasma testing by Labcorp (Laboratory Corporation of America Holdings, Burlington, NC, formerly known as Genetica DNA Laboratories) to confirm their authenticity and integrity.
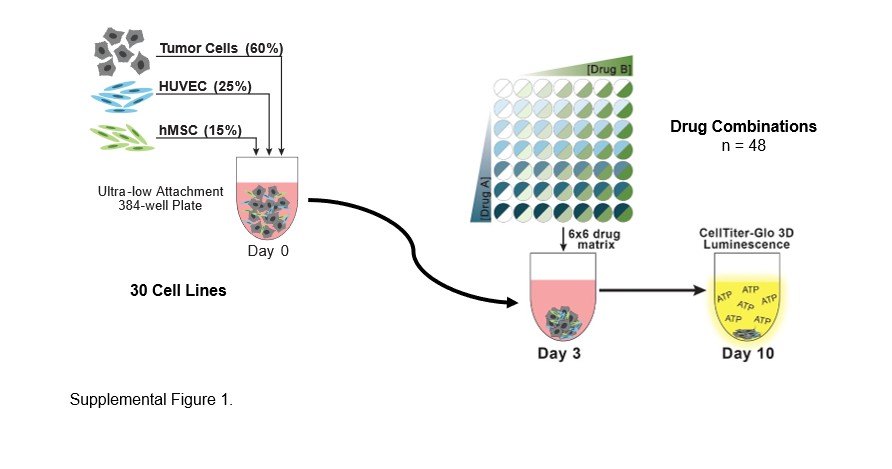
High-throughput Drug Combination Screening.
Prior to their inoculation into microplates, malignant cells, HUVEC, and hMSC were removed from T flasks using TrypLE express (Invitrogen, cat. 12605036) and harvested by centrifugation for 5 min at 233 × g. Following removal of the supernatant, the cells were resuspended in fresh medium and counted using a Cellometer auto T4 bright field cell counter (Nexcelom, Lawrence, MA) and trypan blue to distinguish viable cells. Multi-cell type (mct) tumor spheroids were grown from the mixture of three cell types: 60% malignant cells, 25% HUVECs, and 15% hMSCs as described previously 2]. Mixed cell suspensions of 50 μL were dispensed into the wells of 384-well black/clear round-bottom ULA spheroid microplates (Corning Inc., cat. 3830). Following inoculation, the microplates were transferred to an incubator (Thermo Fisher Scientific, Waltham, MA) and maintained at 37 °C and 5% CO_2_ with 95% humidity. Three days after inoculation, test agents or controls were delivered to the wells of microplates. The approved and investigational anticancer agents, prepared as 800× stock solutions, were subsequently transferred in 62.5 nL volumes to the appropriate wells of microplates using an I.DOT non-contact dispenser (DISPENDIX, Stuttgart, Germany) to achieve a 1x final concentration. All anticancer agents and their combinations were tested in quadruplicate. Additionally, each microplate included a DMSO vehicle control (n = 16) and a cytotoxicity control (1 μM staurosporine and 3 μM gemcitabine, n = 20). After delivery of the test agents and controls, the microplates were returned to the incubator for 7 days. Ten days after inoculation, the assay was completed with the addition of 20 μL CellTiter-Glo 3D (Promega, Madison, WI, cat. G9683) to each well. Next, the microplates were placed on a microplate shaker for 5 min. After 25 min of incubation at room temperature, luminescence was measured as a surrogate indicator of cell viability using a PHERAstar FSX microplate reader (BMG LABTECH, Cary, NC) [21, 22].
Data Analysis.
Luminescence measurements from the screen were exported as comma separated values (CSV) files and imported into custom Excel spreadsheets (Microsoft, Redmond, WA) for analysis. The raw luminescence data were evaluated for quality control, filtered for outliers, and converted to percent viability by normalizing to the DMSO (vehicle-treated) control. Concentration-response data were fit to the four-parameter logistic equation using the Solver Add-In in Excel. The Bliss independence model states that if two drugs have independent activities, then the viability for the combination is equal to the product of the viability of the two single agents [22, 23]. Synergy between two compounds was indicated by a lower observed percent viability than predicted by the Bliss independence model, whereas antagonism was indicated by a greater observed percent viability than predicted. The mean and statistical significance of Bliss independence scores for each drug combination-model across all concentrations and biological replicates were evaluated [23]. Response surface maps were generated using the MATLAB web application where blue indicates synergy and red indicates antagonism.
Data Availability.
All data are accessible via the PubChem BioAssay public database (AID 1918931; AID1918932; AID1918933; AID1918930; AID1918934; AID1918935; AID1918936; AID1918937; AID1918938; AID8939; AID1918942; AID8944; AID1918943; AID1918945; AID1918946; AID1918947; AID1918949; AID1918948; AID8950; AID1918951; AID1918952; AID1918953; AID1918954; AID1918955; AID1918957; AID1918958; AID1918940; AID1918956; AID8941; AID2060627; AID 2060626; AID 2060625; AID 2060624; AID 2060623; AID 2060613; AID 2060622; AID 2060619; AID 2060621; AID 2060620; AID 2060616; AID 2060604; AID 2060603; AID 2060618; AID 2060617; AID 2060615; AID 2060612; AID 2060614; AID 2060610; AID 2060611; AID 2060602; AID 2060599; AID 2060609; AID 2060601; AID 2060608; AID 2060607; AID 2060606; AID 2060605; AID 2060600 (Table 4).
RESULTS
Single agent concentration response data for two investigational CLK inhibitors cirtuvivint and CC-671, and an investigational LSD1 inhibitor iadademstat, are shown in Figure 1. The compounds were tested 26 human tumor cells lines grown as mct-spheroids in 9-point concentration response at concentrations spanning 4-logs with an exposure time of 7 days. Cirtuvivint was the most potent cytotoxin with IC_90_ concentrations between 0.2 to 10 μM. The LSD-1 inhibitor iadademstat was assessed in mct-spheroids in 29 cell lines including PDMR lines and established lines. The most responsive lines were the NCI-H211 SCLC, two triple-negative breast cancer lines MDA-MB-231 and MDA-MB-468, a pancreatic carcinoma line 292921-168-R-J2, and an ovarian carcinoma line 556581-035-R-J1; however, only the NCI-H211 mct-spheroids reached an IC_50_ at 7.5 μM. Iadademstat was the least cytotoxic and reached an IC_50_ concentration only in the MDA-MB-468 cell line at 10 μM, the highest concentration tested. Cirtuvivint, CC-671 and iadademstat were tested in combination with anticancer drugs and investigational agents in complex mct-spheroids composed of human tumor cells with known genetic alterations, HUVEC and hMSC cells. The drugs and investigational agents tested are listed in Table 1. The genetic variants for key genes in the tumor cell lines are shown on Table 2. Drugs and investigational agents were tested at concentrations beginning at the clinical Cmax concentration or at 10 μM, if the clinical Cmax was not established, decreasing in half-log increments for 6 concentrations.
The KRAS G12D selective inhibitor MRTX-1133 was assessed in combination with cirtuvivint in four PDMR lines harboring the KRAS G12D mutation and the 616215-338-R-J1 colon carcinoma which harbors a KRAS G12S mutation (Figure 2) [24]. The PDMR lines with KRAS G12D were among the most responsive to MRTX-1133 as a single agent with 377384-186-R-J1 pancreatic carcinoma mct-spheroids being most responsive. Simultaneous combination of cirtuvivint and MRTX-1133 produced primarily additive cytotoxicity with a few regions of great-than-additive cytotoxicity in the 186277-243-T-J2 colon carcinoma, 996289-038-R-J1 colon carcinoma, 377384-186-R-J1 pancreatic carcinoma and 242566-281-R-J2 pancreatic carcinoma mct-spheroids with up to 2.5–3-logs of cell killing. The 616215-338-R-J1 colon carcinoma harboring a KRAS G12S mutation was also responsive to the combination of cirtuvivint and MTRX-1133. The response of the same five tumor mct-spheroids was assessed after 7-days exposure to CC-671 with MRTX-1133 (Figure 2). Tumor cell killing was greatest in the 377384-186-R-J1 pancreatic carcinoma complex mct-spheroids reaching 2-logs which was additive by the Bliss independence calculation. Greater-than-additive cell killing was evident under the same conditions for the 377384-186-R-J1 pancreatic carcinoma, the186277-243-T-J2 colon carcinoma, 996289-038-R-J1 colon carcinoma mct-spheroids. The maximal tumor cell killing achieved with CC-671 and MRTX-1133 in the 377384-186-R-J1 pancreatic carcinoma, the 186277-243-T-J2 colon carcinoma, 996289-038-R-J1 colon carcinoma and 616215-338-R-J1 colon carcinoma mct-spheroids was 1-log.
Moving down-stream of RAS, the simultaneous combination of cirtuvivint and the pan-PI3K inhibitor copanlisib was examined in five PDMR cell lines, the 186277-243-T-J2 colon carcinoma, the 168753-222-R-J1 bladder carcinoma, 947758-054-R-J2 colon carcinoma, 485368-065-R4-J2 pancreatic carcinoma and 454973-116-R3-J5 pancreatic carcinoma mct-spheroids with a 7-day exposure (Figure 3A) [25]. The 168753-222-R-J1 bladder carcinoma mct-spheroids were more responsive to copanlisib as a single agent than were the 186277-243-T-J2 colon carcinoma mct-spheroids. The tumor cell killing was primarily additive in both tumor lines reaching 2-logs for the 186277-243-T-J2 colon carcinoma mct-spheroids and 3-logs for the168753-222-R-J1 bladder carcinoma mct-spheroids. The 947758-054-R-J2 colon carcinoma, 485368-065-R4-J2 pancreatic carcinoma and 454973-116-R3-J5 pancreatic carcinoma mct-spheroids with a 7-day exposure were similarly responsive to the combination of copanlisib and cirtuvivint with cell killing reaching 2- to −3-logs. The same five PDMR cell lines were exposed to CC-671 in simultaneous combination with copanlisib (Figure 3A). The combination of CC-671 with copanlisib was less cytotoxic than the combination of cirtuvivint with copanlisib in each of the five cell lines tested as mct-spheroids. The combination produced maximal cytotoxicity in the 168753-222-R-J1 bladder carcinoma mct-spheroids with additive to greater-than additive cell killing reaching 2-logs while the same combination produced less than 1-log of cell kiliing in the 485368-065-R4-J2 pancreatic carcinoma mct-spheroids.
The simultaneous combination of cituvivint and the PI3Ka selective inhibitor inavolisib in 186277-243-T-J2 colon carcinoma mct-spheroids resulted in additive to greater-than-additive cytotoxicity reaching a maximum to 2-logs (Figure 3B) [26]. As determined by the Bliss independence calculation the combination of cirtuvivint and inavolisib produced additive cell killing in the 168753-222-RJ1 bladder carcinoma mct-spheroids reaching 3-logs at the 2 highest concentrations of cirtuvivint across the concentration range of inavolisib. Greater-than-additive cell killing was seen with inavolisib in simultaneous combination with CC-671 in mct-spheroids grown from 947758-054-R-J2 colon carcinoma cells and 186277-243-T-J2 colon carcinoma cells (Figure 3B). Neither 947758-054-RJ2 colon carcinoma mct-spheroids nor 186277-243-TJ2 colon carcinoma mct-spheroids was very responsive to CC-671 reaching 62% and 70% cell killing, respectively. The combination of CC-671 and inavolisib reached nearly 2-logs of cell killing in both lines with more evidence of greater-than-additive cell killing occurring in the 947758-;054-R-J2 colon carcinoma mct-spheroids. Inavolisib was not effective in the 947758-054-R-J2 colon carcinoma mct-spheroids; however, greater-than-additive killing occurred over the 6×6 concentration matrix, the same was true of the combination of CC-671 and inavolisib in the 186277-243-TJ2 colon carcinoma mct-spheroids albeit the magnitude was less. Overall, the combination of CC-671 and inavolisib reached 1.5-logs of cell killing in both tumor lines.
As a single agent the XPO1 inhibitor eltanexor, resulted in 1- to 2-logs of cytotoxicity in mct-spheroids after a 7-day exposure [27, 28]. Five cell lines were selected to highlight the results from the combination of cirtuvivint and eltanexor (Figure 4). The simultaneous combination of cirtuvivint with eltanexor produced additive to greater-than-additive killing of 616215-338-R-J1 colon carcinoma mct-spheroids. While greater-than-additive cytotoxicity of the combination was observed at moderate concentrations of eltanexor and lower concentrations of cirtuvivint, cell killing of 2- to 3-logs was observed at the higher concentrations of both agents. The simultaneous combination of cirtuvivint and eltanexor produced greater-than-additive cytotoxicity at moderate concentrations of both agents in the 996289-038-R-J1 colon carcinoma mct-spheroids reaching 2.5-logs. The simultaneous combination of cirtuvivint and eltanexor was additive in the 876135-273-R-J2 melanoma, the 648629-189-R-J1 bladder carcinoma and 377384-186-R-J1 pancreatic carcinoma. Additive to less-than additive tumor cell killing occurred over a wide concentration range of both CC-671 and eltanexor in 616215-338-R-J1 colon carcinoma and the 996289-038-R-J1 colon carcinoma mct-spheroids reaching a maximum of about 1.5-logs. The greatest depth of tumor cell killing was observed in the 876135-273-R-J2 melanoma mct-spheroids reaching 3-logs at the two highest concentrations of CC-671 and eltanexor (Figure 4).
The DNA-PK inhibitor peposertib was studied in combination with the CLK inhibitor cirtuvivint or with the LSD-1 inhibitor iadademstat (Figure 5A) [29–31]. While peposertib as a single agent was not highly effective in the five PDMR cell lines highlighted, 1566681-154-R-J1 melanoma, 233499-124-R-J3 and 616215-338-R-J1 colon carcinomas, 324938-238-R-1 and 565232-114-T-J1 bladder carcinomas, the combination of peposertib and cirtuvivint resulted in 2- to 3-logs of cell killing in each of the 5 lines grown as mct-spheroids. In contrast, the combination of peposertib with the LSD-1 inhibitor iadademstat resulted in sub-additive to additive cell killing in the five PDMR lines highlighted, 186277-243-T-J2 and 233499-124-R-J3 colon carcinomas, 324938-238-R-1 bladder carcinoma, 743899-274-T renal cell carcinoma and 521955-158-R2-J5 pancreatic carcinoma (Figure 5A).
SN-38 is the active metabolite of the prodrug irinotecan, that acts, like irinotecan, by inhibiting topoisomerase I, an enzyme induces a single strand break in DNA to relax the DNA during replication [32, 33]. SN-38 was tested at concentrations up to 1μM during the 7-day exposure period (Figure 5B). At the lower concentrations of cirtuvivint and moderate to high concentrations of SN-38 marked greater-than-additive tumor cell killing with Bliss additivity scores up to 66 for the 616215-338-R-J1 colon carcinoma, 58 for the 233499-124-R-J3 colon carcinoma, 40 for the 565232-114-T-J1 bladder carcinoma, and 21 for the 186277-243-T-J2 colon carcinoma while, the combination of cirtuvivint and SN-38 produced additive cytotoxicity due to the sensitivity of the 186277-243-T-J2 mct-spheroid to SN-38 which killed 1-log of cells as a single agent (Figure 5B). By comparison, the combination of iadademstat and SN-38 produced less-than-additive to additive cell killing over the iadademstat and SN-38 concentrations tested.
The histone deacetylase inhibitor entinostat was tested in combination with cirtuvivint and iadademstat and the results from five PDMR human tumor cell lines grown as mct-spheroids are shown in Figure 6A [34, 35]. The combination of entinostat with cirtuvivint were additive in the five cell lines highlighted over the concentration ranges of the drugs tested with a 7-day exposure time. A similar result was obtained with the combination of iadademstat with entinostat with the five PDMR cells lines grown as mct-spheroids producing less-than-additive to additive cytotoxicity.
Combinations of cirtuvivint and iadadematat with the PARP1 inhibitor olaparib in five of the PDMR tumor cell lines grown as mct-spheroids resulted in the combination producing sub-additive to additive cytotoxic effects in the 324938-238-R-1 and 565232-114-T-J1 bladder carcinomas and additive to greater-than additive cytotoxicity in the 186277-243-T-J2 and 616215-338-R-J1 colon carcinomas (Figure 6B)[36]. The combination of iadademstat with olaparib resulted in little cytotoxicity with combinations just reaching an IC_50_ with the highest concentration of both drugs.
Iadademstat was tested with the NEDD8-activating enzyme (NAE) inhibitor pevonedistat (Figure 7). Pevonedistat prevents the activation of cullin-RING E3 ligases thereby blocking the ubiquitination and proteasomal degradation of cellular proteins causing a build-up of proteins leading to cell death [37–41]. The K-562 leukemia was responsive to pevonedistat reaching 1 log of cytotoxicity at the highest concentration (3 mM) tested. There was an iadademstat concentration dependent increase in cytotoxicity with iadademstat and pevonedistat reaching 2-logs at 3 mM pevonedistat (Figure 7). Of the two PDMR pancreatic carcinoma lines, the less responsive the 521955-158-R2-J5 pancreatic carcinoma mct-spheroids reached >1-log, while the 292921-186-R-J2 pancreatic carcinoma mct-spheroids reached 2-logs of cell killing at higher pevonedistat concentrations. The OVCAR-5, and NCI/ADR-RES ovarian carcinoma mct- spheroids were responsive to pevonedistat reaching 1-log at 3mM pevonedistat. Combination of pevonedistat with iadademstat did not increase OVCAR-5 or NCI/ADR-RES mct-spheroid killing compared with pevonedistat alone.
TAK-243 is an inhibitor of the (UAE). UAE is the primary E1 enzyme regulating the ubiquitin conjugation cascade [42–43]. The binding of TAK-243 to UAE prevents protein ubiquitination resulting in protein accumulation (proteotoxic stress) and leading to cell death. The K-562 leukemia was responsive to TAK-243 with 3-logs of cytotoxicity at the highest concentration of TAK-243 (0.3 mM) tested (Figure 7). Iadademstat with TAK-243 resulted in an iadademstat concentration dependent increase in K-562 leukemia cell cytotoxicity at the lower concentrations of TAK-243 compared with TAK-243 alone. Two of the PDMR pancreatic carcinoma, 292921-186-R-J2 and 521955-158-R2-J5, were as responsive to TAK-243 as was the K-562 leukemia reaching 3-logs of cytotoxicity. Iadademstat with TAK-243 increase in cytotoxicity in the two pancreatic carcinoma mct-spheroids compared with TAK-243. TAK-243 and iadademstat produced a modest increase in cytotoxicity. The PDMR 556581-035-R-J1 ovarian carcinoma was highly responsive to TAK-243 reaching 3-logs of cytotoxicity. Iadademstat with TAK-243 increased cytotoxicity compared with TAK-243 alone at the mid-range concentrations. The NCI/ADR-RES ovarian carcinoma mct-spheroids were responsive to TAK-243 reaching 2-logs of cytotoxicity. Iadademstat with TAK-243 was less cytotoxic than TAK-243 as a single agent in the NCI/ADR-RES cell line. The OVCAR-5 cell line was responsive to TAK-243 reaching nearly 2-logs of cell killing. The combination of iadademstat and TAK-243 was modestly more cytotoxic than TAK-243.
DISCUSSION
Most genes with multiple introns and exons undergo alternative splicing to generate multiple mRNAs which are then translated into the diversity of proteins making up cellular proteins required for differentiation, development, and cell death in a process performed by spliceosomes, highly complex structures made up of approximately 300 proteins and RNA [45–47]. The Cancer Genome Atlas (TCGA) indicates that many solid tumors and hematologic malignancies deregulate DYRK1A. The pan-CLK/DYRK inhibitor cirtuvivint can cause programmed cell death at concentrations which inhibit the accumulation of phosphorylated SR proteins and alter splicing decreasing cellular proliferation in hematologic PDXs [45]. Cirtuvivint inhibits spliceosome associated CLK kinases especially SRSF5/6, thus, providing indirect inhibition of Wnt signaling resulting in antitumor activity [46]. Approximately 90% of colorectal cancers have mutations in the Wnt/β-catenin signaling pathway and aberrant Wnt signaling often occurs in gastric, pancreatic, breast and other cancers [46]. CC-671 is a potent and selective inhibitor of both TTK (human protein kinase monopolar spindle 1 [hMps1]) and CDC like kinase 2 (CLK2). A protein docking analysis indicated that CC-671 has high binding affinity to the drug-binding site of ABCG2 [47]. While dysregulation and alteration in premRNA splicing is a recognized therapeutic target in hematologic malignancies, targeting pre-mRNA splicing has only been pursued recently for solid tumors. Pre-mRNA splicing modulation followed by PARP inhibition or chemotherapy in BRCA-mutant breast and ovarian cancers characterized by a “BRCA-ness” phenotype of dysfunctional homologous DNA repair [48].
Spliceosome-associated SR protein kinases SRPKs, CLKs, and NEK2 are altered in many cancers [49]. The CLK kinase family which includes CLK1-4, in conjunction with SRPK kinases adjust phosphorylation of SR proteins to modulate alternative splicing. CLK kinase activity changes are associated with cancer development and progression. Phosphorylation of SR proteins impact their subcellular localization, association with the spliceosome complex, and splicing activity[49].SRSF5 and SRSF3 were reported to be overexpressed in oral squamous cell carcinoma (OSCC), and necessary for OSCC cell proliferation, cell cycle progression, and in vivo tumor formation. SRSF5-7 were found to be upregulated in small cell lung cancer (SCLC) and NSCLC tissues, and knockdown of SRSF5-7 in SCLC cell lines showed a significant decrease in proliferation [49].
Epigenetic mechanisms control gene expression patterns without change in DNA sequence. Histones, DNA binding proteins involved in regulation of nucleosome function, are subject to methylation, acetylation, phosphorylation, and ubiquitination. The methylation and demethylation of lysine residues on histone tails are post-translational protein modifications which control gene expression. The KDM (K=lysine) demethylase gene family includes 20 KDMs. KDM1s utilize a flavin adenine dinucleotide cofactor to demethylate methylated lysine substrates. KDM1A regulates many aspects of cell biology including self-renewal, differentiation, and stem cell pluripotency [50]. The protein encoded by the KDM1A gene is LSD1 which removes mono- and dimethyl groups from histone H34K and other chromatin-associated proteins. Two ways epigenetic therapies act as anticancer therapies are repression of oncogene function or activation of tumor-suppressor genes [51]. LSD1 is overexpressed in many proliferative diseases including hematological, lung, breast, and prostate cancers. Iadademstat, an irreversible LSD1 inhibitor, is in clinical development [52–54]. Several LSD1 inhibitors have completed Phase 1 clinical trial and have moved on to Phase 2 studies [55, 56]. Pevonedistat interferes with the function of the proteasome pathway blocking NAE. The PDMR 292921-168-R-J2 pancreatic carcinoma mct-spheroids was most responsive to pevonedistat reaching 2-logs of cytotoxicity and the K-562 leukemia, the PDMR 521955-158-R-J5 pancreatic carcinoma and the ovarian carcinoma lines OVCAR-5 and NCI/ADR-RES mct-spheroids reached 1-log of cytotoxicity upon exposure to pevonedistat for 7 days. The combination of iadademstat and pevonedistat resulted in increased cytotoxicity in the K-562 leukemia and the PDMR 556581-035-R-J1 ovarian carcinoma compared with single agent pevonedistat (Figure 7). TAK-243 was markedly cytotoxic as a single agent in the K-562 leukemia, in 2 of 3 PDMR pancreatic carcinomas, 292921-168-R-J2 and 521955-158-R-J5, and the PDMR 556581-035-R-J1 ovarian carcinoma resulting in 3-logs of cytotoxicity at the highest concentration (0.3 mM) tested. There was little increase in response with iadademstat and TAK-243.
A first-in-human phase 1 iadademstat clinical study was conducted in relapsed refractory acute leukemia enriched with MLL/KMT2A-rearranged acute myeloid leukemia patients with most having MLL-translocation disease. The pharmacokinetic data indicate that the iadademstat clinical Cmax concentration was 0.116–0.182 nM, a concentration below the concentration range of 0.1–10 μM in the current study [57, 58]. Some patients with a molecular response to treatment with iadademstat showed blast cell differentiation, but no clinical responses were seen. In the current screen of 29 cell lines and 25 drugs and investigational agents indicate that response is highly cell line dependent. Although genetic marker(s) that might correlate with response to the combination regimens were sought no clear marker(s) emerged.
Supplementary Files
This is a list of supplementary files associated with this preprint. Click to download.
{kind=link}
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Bradley RK, Anczuków O. RNA splicing dysregulation and the hallmarks of cancer. Nature Rev Cancer 2023; 23: 135–55.36627445 10.1038/s 41568-022-00541-7PMC 10132032 · doi ↗ · pubmed ↗
- 2Aubol BE, Wozniak JM, Fattet L, Gonzalez DJ, Adams JA. CLK 1 reorganizes the splicing factor U 1–70K for early spliceosomal protein assembly. Proc Natl Acad Sci 2021; 118: e 2018251118.33811140 10.1073/pnas.2018251118 PMC 8040622 · doi ↗ · pubmed ↗
- 3Yoshimi A, Abdel-Wahab O. Molecular pathways: understanding and targeting mutant spliceosomal proteins. Clin Cancer Res 2017; 23: 336–41.27836865 10.1158/1078-0432.CCR-16-0131 PMC 5241248 · doi ↗ · pubmed ↗
- 4Moyano PM, Nemec V, Paruch K. Cdc-like kinases (CL Ks): biology, chemical probes, and therapeutic potential. Int J Mol Sci 2020; 21: 7549.33066143 10.3390/ijms 21207549 PMC 7593917 · doi ↗ · pubmed ↗
- 5Song M, Pang L, Zhang M, Qu Y, Laster KV, Dong Z. Cdc 2-like kinases: structure, biological function, and therapeutic targets for diseases. Sig Transduc Targ Therap 2023; 8:148.10.1038/s 41392-023-01409-4PMC 1008206937029108 · doi ↗ · pubmed ↗
- 6Corr BR, Moroney MR, Woodruff E, Watson ZL, Jordan KR, Danhorn T, Bailey C, Wolsky RJ, Bitler BG. Combination CDC-like kinase inhibition (CLK)/Dual-specificity tyrosine-regulated kinase (DYRK) and taxane therapy in CTNNB 1-mutated endometrial cancer. bio Rxiv: posted April 6, 2023.
- 7Riggs JR, Nagy M, Elsner J, Erdman P, Cashion D, Robinson D, Harris R, Huang D, Tehrani L, Deyanat-Yazdi G, Narla RK, Peng X, Tran T, Barnes L, Miller T, Katz J, Tang Y, Chen M, Moghaddam MF, Bahmanyar S, Pagarigan B, Delker S, Le Brun L, Chamberlain PP, Calabrese A, Canan SS, Leftheris K, Zhu D, Boylan JF. The discovery of a dual TTK protein kinase/CDC 2-like kinase (CLK 2) inhibitor for the treatment of triple negative breast cancer initiated from a phenotypic screen. J Med Chem 2017; 60: 8989–9002.28991472 10.1021/acs.jmedc · doi ↗ · pubmed ↗
- 8Zhu D, Xu S, Deyanat-Yazdi G, Peng SX, Barnes LA, Narla RK, Tran T, Mikolon D, Ning Y, Shi T, Jiang N, Raymon HK, Riggs JR, Boylan JF. Synthetic lethal strategy identifies a potent and selective TTK and CLK 1/2 inhibitor for treatment of triple-negative breast cancer with a compromised G 1–S checkpoint. Mol Cancer Ther 2018; 1: 17: 27–38.10.1158/1535-7163.MCT-17-108429866747 · doi ↗ · pubmed ↗