First molecular detection and multilocus genotyping of Enterocytozoon bieneusi from pigs in Guangxi Zhuang Autonomous region, Southern China
Si-Ang Li, Yang Liu, Hui-Hong Lu, Ya-Fei Song, Meng-Jie Chu, Fei Huang, Shu-Yan Wang, Dong-Hui Zhou

TL;DR
This study found that a parasite called Enterocytozoon bieneusi infects pigs in southern China and identified new types that can potentially spread to humans.
Contribution
First molecular detection and multilocus genotyping of E. bieneusi in pigs in Guangxi, China, revealing zoonotic potential.
Findings
24.55% of pig fecal samples tested positive for E. bieneusi, with significant regional and farming differences.
Ten novel genotypes (GXP-1 to GXP-10) and 12 known genotypes were identified, all belonging to the zoonotic group 1.
44 distinct multilocus genotypes were observed, indicating high genetic diversity and potential for human transmission.
Abstract
is a cosmopolitan microsporidian that infects a wide range of vertebrate and invertebrate hosts including humans, domestic animals and wild game. In this study, we determined the prevalence of E. bieneusi in pigs from Guangxi Zhuang Autonomous Region, China, examined the different genotypes present, and assessed their zoonotic potential. This study investigate the prevalence and multilocus genotyping of E. bieneusi in pigs from Guangxi Zhuang Autonomous Region in China. We collected 721 fecal samples from pigs in four regions (Guigang, Nanning, Hezhou and Yulin). These samples were subsequently analyzed using nested PCR and multilocus sequence typing (MLST). The results demonstrated that the overall prevalence of E. bieneusi in pigs was 24.55%, ranging from 11.48 to 43.26% among four regions. The infection rates of E. bieneusi in pigs of four types (breeding pigs, piglets, nursery…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
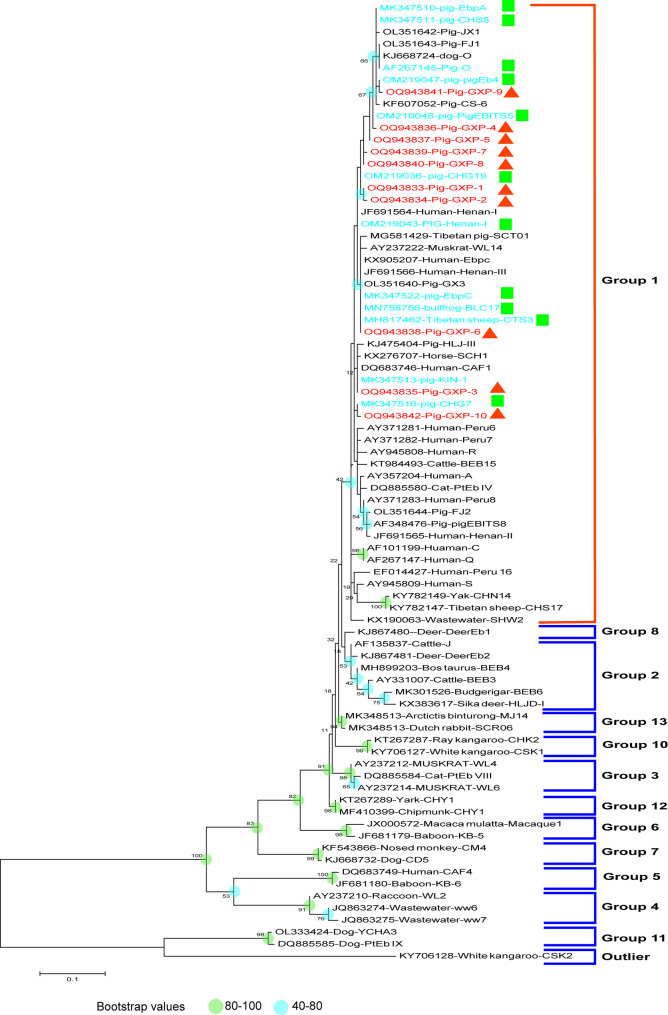 Figure 1
Figure 1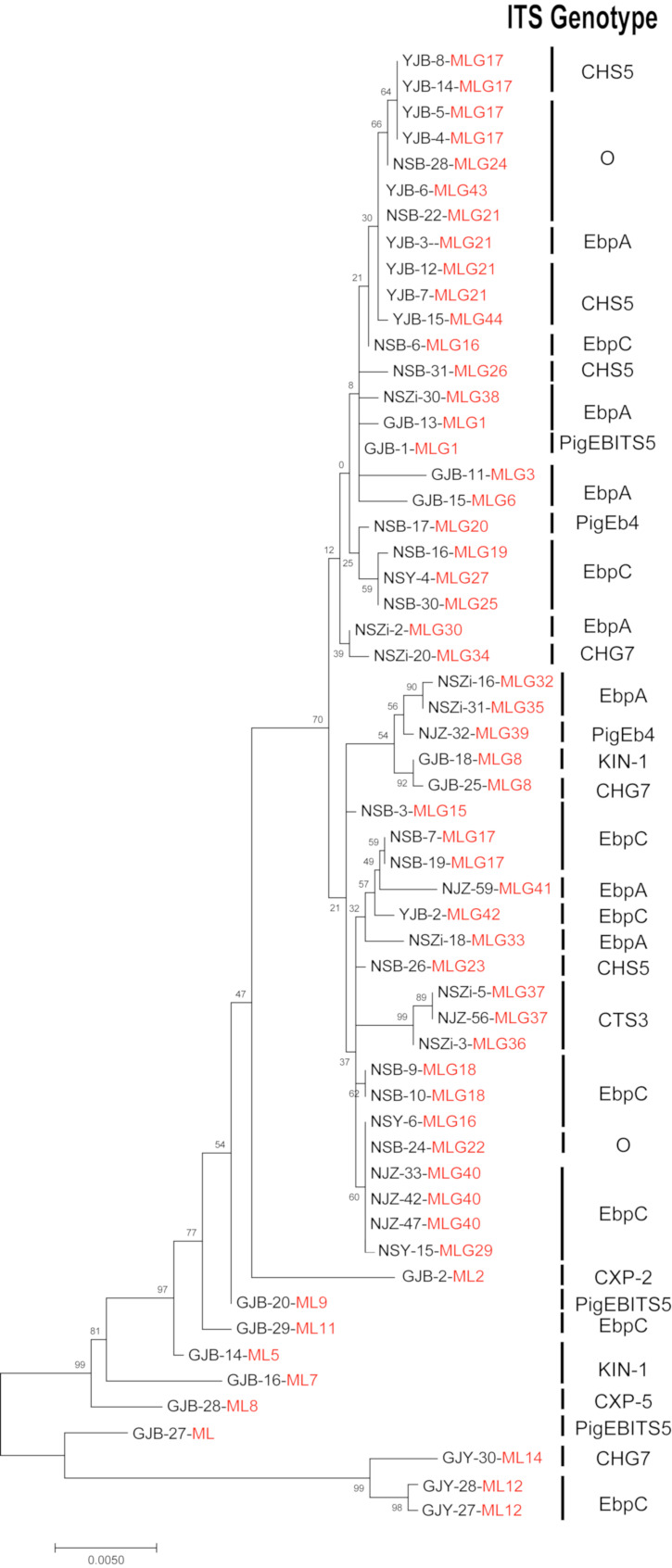 Figure 2
Figure 2- —Guangxi Natural Science Foundation
- —Natural Science Foundation of Fujian Province of China
- —Industry-university-research Cooperation Project in Fujian Province University and enterprise
- —Research Foundation of Engineering Research Center for the Prevention and Control of Animal Original Zoonosis, Fujian Province University
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsParasitic Infections and Diagnostics · Plant and Fungal Interactions Research · Toxoplasma gondii Research Studies
Background
Microsporidia are unicellular eukaryotes that exclusively parasitize the intestinal cells of their hosts and can infect a diverse range of animals, including humans, domestic animals, wild animals and companion animals [1–4]. To date, microsporidia have been classified into more than 220 genera and 1,700 species, among which 17 microsporidia species have been demonstrated to be capable of infecting humans [2, 5]. Among these species, Enterocytozoon bieneusi is the most widely reported opportunistic pathogen, causing more than 90% of microsporidiosis in human [6–8]. E. bieneusi is generally infected with weakened immune system individuals, such as HIV/AIDS-infected patients, organ transplant recipients, the elderly and children [7, 9–11]. Reported studies have shown that E. bieneusi can widely infect with various important economic animals, including pigs, dairy cows, goats and sheep. The highest prevalence of E. bieneusi in animals occurs in pigs (60%), followed by goats (19.2%) and cattle (5%), whereas the prevalence of E. bieneusi in humans was detected in only 1% [12]. The main transmitted route of E. bieneusi is fecal-oral transmission. Humans or other animals become infected by ingesting food or water contaminated by spores of E. bieneusi [9, 13–15]. However, other routes, such as respiratory transmission via inhalation of E. bieneusi spores also contribute to the spread of this pathogen [16]. A previous study reported that individuals occupationally or non-occupationally exposed to urban feral pigeons for 30 min may inhale approximately 3.5 × 10³ E. bieneusi spores, while nearby individuals may inhale around 1.3 × 10³ spores, leading to E. bieneusi infection [17]. The proliferation of mature spores in the intestinal epithelial cells, leads to shortening and reduction of surface area of the intestinal villus, causing malabsorption in the host [18, 19]. In recent years, prevalence surveys and genotype identification of E. bieneusi in pigs have been conducted in many regions of the world for assessing the risks for its zoonotic potential [20]. Previously reported studies have shown that pigs infected with E. bieneusi were usually asymptomatic, but the early pigs infected with E. bieneusi can excrete the spores of E. bieneusi for life which can contaminate the environment [20–22].
The spores of E. bieneusi, measured only 1.1 to 1.6 by 0.7 to 1.0 μm, are exceptionally tiny, so it is a challenge to distinguish them under light microscopes during fecal detection [18]. The detection sensitivity of the electron microscopy is low and prone to false negative result [23]. In addition, Microscopy with Gram chromotrope or modified trichrome staining is the standard method for diagnosing microsporidiosis [24]. Microsporidia spores stained with these techniques appear dark purple (Gram chromotrope) or bright red (modified trichrome) [25]. However, these staining methods are not only time-consuming and the fixatives are toxic (containing mercury and chlorine), but also unable to distinguish the species of microsporidia, with the sensitivity for fecal samples limited to 64% [23, 26]. In recent years, Polymerase Chain Reaction (PCR) targeting the internal transcribed spacer (ITS) region of the small subunits of ribosomal rRNA (SSU rRNA) gene has been widely adopted for genotyping E. bieneusi. This highly polymorphic locus enables precise discrimination of morphologically indistinguishable isolates [27, 28]. The use of sensitive and discriminative molecular technique will contribute to determining the true prevalence of microsporidiosis and possibly their potential transmission source depending on species identification [28]. To date, at least 600 genotypes have been identified based on ITS gene of E. bieneusi, which were classified into 11 different groups (group 1–11) [29]. Currently, more than 30 studies on E. bieneusi in pigs have been published from 14 countries, and more than 130 ITS genotypes of E. bieneusi have been identified in pigs and wild boars worldwide [30]. Among the genotypes detected in pigs, 19 genotypes of them have also been detected in human samples (CHN1, Bfrmr2, CAF1, CS-1, CS-4, D, EbpA, EbpC, EbpD, H, Henan-III, Henan-IV, I, LW1, O, PigEBITS5, PigEBITS7, PigEB10, SH8) [30]. Nevertheless, the use of single marker genes alone has inherent limitations in E. bieneusi genotyping. In 2011, a multilocus sequence typing (MLST) tool offering a high-resolution for genotyping E. bieneusi was found by Feng et al. based on microsatellite (MS1, MS3 and MS7) and minisatellite (MS4) loci. The tool was used to explore genotype taxonomy and host specificity of E. bieneusi [31].
Since the first report of E. bieneusi infection in pigs in 1996 [1], similar reports have been continuously documented worldwide. So far, E. bieneusi has been detected from pigs in a number of countries, including China, Malaysia, Czech and Spain [20, 22, 32, 33]. While E. bieneusi infection has been reported in several Chinese provinces, including infection rates of 79.8% in Southwest China, 37.5% in Zhijiang, and 46.8% in Hainan [30, 34, 35], there have been few studies on the prevalence of E. bieneusi in pigs from Guangxi Zhuang Autonomous Region. To address this research gap, this study aims to comprehensively investigate the prevalence and genotype distribution of E. bieneusi from pig populations in Guangxi Zhuang Autonomous Region. Through using nested PCR technology and multilocus sequence typing (MLST) analysis to identified and characterized for E. bieneusi. Ultimately, we evaluate the risk of zoonotic transmission of E. bieneusi by phylogenetic analysis. These results help to expand the knowledge of E. bieneusi distribution and provide baseline data for the prevention and control of E. bieneusi in China.
Materials and methods
Study sampling
The pig’s population in the Guangxi Zhuang Autonomous Region reached 41.1 million in 2020. This study, conducted March 2021 to May 2022, a total of 721 fecal samples were collected from free-range pig farms (314) and intensive pig farms (407) in four regions of Guangxi Zhuang Autonomous Region (177 samples from Guigang, 215 samples from Nanning, 183 samples from Hezhou and 146 samples from Yulin). These samples included 175 from breeding pigs, 164 from fattening pigs, 198 from nursery pigs and 184 from piglets. All fecal samples were directly collected from each pig’s rectum or were immediately collected from the fresh feces on the ground after defecation using sterile polyethylene gloves. These samples were numbered and marked with the information about the regions, types and feeding modes of the pig, and then stored at -80 °C until DNA extraction.
Isolation of genomic DNA
Pretreatment of samples before DNA extraction: all fecal samples were heated at 100 °C for 5 min and then frozen at -80 °C for 5 min [36]. The procedure was repeated five times. According to the instructions of the commercial E.Z.N.A^®^Stool DNA Kit (Omega Biotek Inc., Norcross, GA, USA), the genomic DNA was extracted from processed samples and then stored at 4 °C for further use.
PCR amplification and MLST analysis
E. bieneusi was identified via nested PCR amplification of the ITS region of the rRNA gene with specific primers [27]. The primary PCR amplification using outer primers produced a 435-bp product, while the nested PCR with inner primers yielded a 390-bp product. Detailed PCR reaction conditions and primer sequences for both rounds of amplification are provided in Table S1. Next, ITS positive E. bieneusi strain samples were subjected to nested PCR for amplifying microsatellite and minisatellite targets. Nested PCR targeting the MS1, MS3, MS4, and MS7 loci generated amplicons of 675 bp (MS1), 537 bp (MS3), 885 bp (MS4), and 471 bp (MS7), respectively (Table S1) [31]. All primers were synthesized by Shangya Biotech Co., Ltd. (Fuzhou, China). A positive control (pig-derived DNA of genotype EbpC) and a negative control (reagent water without DNA) were used in all the PCR tests performed to ensure the reliability of the results. The secondary PCR products were separated using 2% agarose gel with ethidium bromide, and then visualized under UV light.
Nucleotide sequencing and phylogenetic analysis
The secondary PCR products of E. bieneusi-positive samples were bidirectionally sequenced by Sangon Biotech Co., Ltd (Xiamen, China). The SeqMan in Lasergene 7.1 software package (DNASTAR Inc., USA) was used to align and assemble bidirectional sequencing results, and then used to check the accuracy of the sequencing results according to the sequence chromatogram for each strand. The processed sequences were compared online with GenBank database using Basic Local Alignment Search Tool (BLAST) (https://blast.ncbi.nlm.nih.gov) for identifying E. bieneusi genotypes. The phylogenetic relationship based on ITS sequences of E. bieneusi was established using 69 E. bieneusi ITS gene reference sequences with the Maximum Likelihood (ML) method under the Kimura 2 parameter model in the software MEGA 11 [37]. Further, we studied the genetic diversity of the clinical strains of E. bieneusi by analyzing 1 minisatellite (MS4) and 3 microsatellites (MS1, MS3, and MS7) according to previously described [31]. Then, we performed a multilocus analysis by combining the four MS sequences to define the multilocus genotypes (MLGs). Sequence alignment was performed using the MegAlign program (DNAstar 7.1, DNASTAR Inc., Madison, WI). And we obtained the tandem sequences of MS1, MS3, MS4, and MS7 through the “Concatenate Sequence” function in Phylosite version 1.2.3 software. Finally, we conducted a phylogenetic analysis of the combined nucleotide sequences by the Kimura-2 parameter model in MEGA 11 software using the Maximum likelihood method. The bootstrap value was set to 1000 for determining support for the clades of the phylogenetic tree. The representative nucleotide sequences obtained in this study were deposited in the GenBank database with accession numbers OQ943833 to OQ943842.
Statistical analysis
SPSS 26.0 (IBM Corp., New York, NY) was used to perform statistical analysis on all data in this study. The Chi-square test was used to assess the associations between infection rates of E. bieneusi and individual husbandry parameters (regions, types and feeding modes). Using the binary logistic regression analysis, the odds ratio (OR) calculated with 95% confidence intervals (95% CI) was explored to measure the strength of association between E. bieneusi prevalence and each univariate factor (regions, types, and feeding modes) [38, 39]. Differences were considered statistically significant when the P-value was less than 0.05.
Results
Prevalence of E. bieneusi in pigs and risk factors
In this study, the overall prevalence of E. bieneusi from pigs in Guangxi Zhuang Autonomous Region was 24.55% (177/721) (Table 1). The infection rate of E. bieneusi in Nanning (43.26%; 93/215) was the highest, followed by Guigang (23.73%; 42/177), Hezhou (11.48%; 21/183) and Yulin (14.38%; 21/146). Infection rate was more likely to be associated with different regions, as indicated by a significant difference in prevalence (χ^2^ = 65.716, P < 0.05). E. bieneusi prevalence in different types of pigs ranged from 9.71 to 42.42%. The highest infection rates of E. bieneusi were found in nursery pigs (42.42%; 84/198), however, the lowest infection rates of E. bieneusi were observed in breeding pigs (9.71%; 17/175). The results showed that the infection rate was more likely to be associated with different growth stages of pigs, and a significant difference was observed among groups (χ^2^ = 60.131, P < 0.05). A statistically significant association was found between feeding mode and E. bieneusi prevalence (χ² = 32.000, P < 0.05), with pigs in free-range farming (34.71%; 109/314) showing higher infection rates than those in intensive farming (16.71%; 68/407). There were significant associations of E. bieneusi-positivity with regions, types and feeding modes of pigs. The highest and lowest prevalence of E. bieneusi were in Nanning (93/215; 43.26%) and Hezhou (21/183; 11.48%), respectively (OR = 1.919; 95% CI [1.157–3.183]). The infection rates of E. bieneusi in nursery pigs were significantly higher than the infection rates of E. bieneusi in fattening pigs (OR = 4.045; 95% CI [2.362–6.926]; P < 0.05). Additionally, free-range farming had 2.897 times higher risk of E. bieneusi infection in pigs than intensive farming (OR = 2.897; 95% CI [1.915–4.382]).
Table 1. Association analysis between risk factors (regions, types and feeding modes) and Enterocytozoon bieneusi test-positivity in this studyRisk factorType of sampleNo. of sampleNo. of the positive sample (%)odds ratio (95% CI)P-valueRegionsHezhou18321 (11.48%)0.323(0.171–0.608)< 0.05Nanning21593 (43.26%)1.919(1.157–3.183)Yulin14621 (14.38%)0.438(0.227–0.847)Guigang *17742 (23.73%)-TypesBreeding pigs17517 (9.71%)0.637(0.320–1.269)< 0.05Piglets18448 (26.09%)1.874(1.070–3.283)Nursery pigs19884 (42.42%)4.045(2.362–6.926)Fattening pigs *16428 (17.07%)-Feeding modesFree-range farming314109 (34.71%)2.897(1.915–4.382)< 0.05Intensive farming 40768 (16.71%)-Total721177 (24.55%) The values were used as references when odds ratio was calculated- Odds ratio was not available
Genotype distribution of E. bieneusi
In this study, based on the ITS gene, 22 genotypes were identified among the 177 *E. bieneusi-*positive samples, including 12 known genotypes and 10 novel genotypes (GXP-1 to GXP-10) (Table 2). Of these identified 12 known genotypes, genotype EbpC (49/177, 27.68%) and EbpA (36/177, 20.34%) were the main dominant genotypes in this study, followed by CHS5 (18/177, 10.17%), PigEb4 (18/177, 10.17%), CHG19 (14/177, 7.91%), CTS3 (13/177, 7.34%), PigEBITS5 (12/177, 6.78%), CHG7 (10/177, 5.65%), KIN-1 (6/177, 3.39%), O (6/177, 3.39%), Henan-I (2/177, 1.13%) and BLC17 (1/177, 0.57%). The novel genotypes with the highest prevalence of E. bieneusi were GXP-2 (11/177, 6.21%), followed by GPX-1 (4/177, 2.26%), GXP-5 (4/177, 2.26%), GXP-7 (3/177, 1.70%) and GXP-6 (2/177, 1.23%). For the novel genotypes GPX-3, GPX-4, GPX-8, GPX-9 and GPX-10, the prevalence of each was 0.57% (1/177).
Table 2. Prevalence and genotypes of Enterocytozoon bieneusi in pigs from Guangxi Zhuang autonomous region, ChinaRisk factorType of sampleNo. of the positive samplePrevalence (%)Genotype (number)RegionsHezhou(21/183)11.48CHG19(12), EbpC(5), CHG7(2), PigEBITS5(1), EbpA(1)Nanning(93/215)43.26EbpA(21), EbpC(25), PigEb4(13), CHS5(12), CTS3(10), PigEBITS5(4), CHG7(3), GXP-5(3), O(3), GXP-7(3), CHG19(2), Henan-I(2), GXP-1(1), GXP-2(1), GXP-4(1), GXP-6(1), GXP-8(1), GXP-9(1), GXP-10(1)Yulin(21/146)14.38EbpC(9), CHS5(6),O(3), EbpA(2),BLC17(1), GXP-1(1)Guiganga(42/177)23.73EbPA(11), EbpC(9), GXP-2(8), KIN-1(5), CTS3(2), PigEBITS5(2), CHG7(4), CHS5(1), GXP-1(1), GXP-3(1), GXP-5(1)TypesBreeding pigs(17/175)9.71CTS3(7), EbpC(5), PigEb4(2), BLC17(1), GXP-7(1), GXP-9(1), GXP-10(1)Piglets(48/184)26.10EbpA(11), CHG19(7), PigEb4(5), EbpC(3), Henan-I(2), CTS3(2), GXP-7(2), CHG7(1), CHG19(1), GXP-4(1), GXP-6(1), GXP-5(1)Nursery pigs(84/198)42.42EbpC(21), EbpA(16), CHS5(15), GXP-2(11), PigEb4(7), O(6), PigEBITS5(6), KIN-1(6), CHG7(4), CTS3(3), GXP-7(2), GXP-1(2), GXP-3(1), GXP-5(1), GXP-5(1)Fattening pigs(28/164)17.10EbpC(16), EbpA(3), CHG7(3), PigEBITS5(2), CHG19(2), CHS5(2), PigEb4(1), GXP-1(1)Feeding modesFree-range farming(109/314)34.71EbpC(29), EbpA(25), CHG19(14), CTS3(12), PigEb4(12), CHS5(11), PigEBITS5(5), CHG7(3), O(3), GXP-5(3), GXP-7(3) Henan-I(2), GXP-6(2), GXP-4(1), GXP-1(1), GXP-2(1),Intensive farming(68/407)16.71EbpC(19), EbpA(11), GXP-2(10), CHG7(7), KIN-1(6), PigEBITS5(6), CHS5(7), O(3), PigEb4(5), GXP-1(2), BLC7(1), CHG19(1), CTS3(1), GXP-3(1), GXP-5(1), GXP-8(1), GXP-9(1), GXP-10(1)Total17735 (19.77%)- There was no genotypic of mixed infections in the sample type
Phylogenetic relationship of E. bieneusi
In this study, we compared the sequences of the identified novel genotypes of E. bieneusi with known sequences and base substitutions (G→T, G→A, A→G, C→T and T→C) at single nucleotide loci (Table 3). Single nucleotide polymorphisms (SNPs) were identified by comparing the ITS gene sequences of the 10 novel genotypes (GXP-1 to GXP-10) with GenBank database, and sequence alignment revealed similarities of was all over 99.47% for all novel genotypes. Compared with the known sequence of EbpC, GXP-1 had a G→T substitution at position 171. Similarly, GXP-2 also had a G→T substitution at position 171 and a G→A substitution at position 212. In comparison with the known sequence of KIN-1, GXP-3 had an A→G substitution at position 219. GXP-4 had a T→C substitution at position 157 compared with its homologous sequence PigEBITS5. Meanwhile, GXP-5 had an A→G substitution at position 212, and GXP-7 had a T→C substitution at position 212 and a G→A substitution at position 219. The homologous sequence of GXP-6 was CTS-3, with a homology of 99.74%, yet it had a G→T substitution at position 171. GXP-8 had the highest homology (99.74%) with the CHG19 sequence, and the substitution occurred at position 157 C→T. Compared with the known sequence of PigEB4, GXP-9 had a T→C substitution at position 251. For GXP-10, compared with its homologous sequence of CHG7, the nucleotide substitution occurred at position 164 G→A. These novel genotypes, although highly similar to the known ones in terms of sequence similarity, still exhibited some minor differences that could be crucial for understanding the genetic diversity of E. bieneusi. Additionally, phylogenetic analysis results indicates that these novel genotypes and 12 known genotypes (EbpC, EbpA, PigEb4, Henan-1, PigEBITS5, CHS5, CTS3, CHG7, CHG19, O, KIN-1, BLC7) identified in this study were clustered into zoonotic group 1 (Fig. 1). In the phylogenetic tree, the novel genotypes were closely clustered with some of the known genotypes, such as EbpC and PigEBITS5, suggesting a relatively close genetic relationship. The presence of novel genotypes within this group also implies that E. bieneusi is still evolving, and these new genotypes may represent new evolutionary directions. Epidemiologically, the fact that these genotypes belong to a zoonotic group means that there is a potential risk of cross-species transmission.
Table 3. Variation in nucleotide loci in the ITS sequence of E. BieneusiGenotypeNucleotide at Position (ITS)Homology ratioGenbank No.107157164171212217219251272344EbpC *GCGGGCATAAMK347522GXP-1---T------99.74%OQ943833GXP-2---TA-----99.47%OQ943834KIN-1 *GCGGGTATAAMK347513GXP-3------G---99.74%OQ943835PigEBITS5 *ATGTATGTAGOM219048GXP-4-C--------99.74%OQ943836GXP-5----G-----99.74%OQ943837GXP-7-----CA---99.47%OQ943839CTS-3 *GCGGGCATAGMH817462GXP-6---T------99.74%OQ943838CHG19 *ACGTGCATATMH817463GXP-8-T--------99.74%OQ943840PigEB4 *ATATATGTGGOM219047GXP-9-------C--99.74%OQ943841CHG7 ACGGGTGTAGMK347516GXP-10--A-------99.74%OQ943842 The genotypes were used as reference when the sequences were compared in multiple sequence comparisons- The nucleotide sites were identical
Fig. 1. Evolutionary relationships of E. bieneusi groups. The relationship between the E. bieneusi genotypes identified in this study and other known genotypes deposited in GenBank was inferred by neighbor-joining analysis of ITS sequences based on genetic distance using the Kimura 2 parameter model. The percentage of replicate trees in which the associated taxa clustered together in the bootstrap test (1000 replicates) is shown next to the branches. The tree is drawn to scale, with branch lengths in the same units as those of the evolutionary distances used to infer the phylogenetic tree. Squares represent the known genotypes, while triangles represent the novel genotypes identified in this study
Multilocus sequence typing of E. bieneusi
To further explore the genetic variations of E. bieneusi isolates from pigs in Guangxi Zhuang Autonomous Region, a total of 177 E. bieneusi-positive samples were studied using the MLST technique. Out of these samples, only 57 samples successfully amplified three microsatellite loci (MS1, MS3, and MS7) and one minisatellite locus (MS4), 44 distinct MLGs (File S1 and Fig S1). The remaining samples could not amplify all four loci. There were 19, 10, 19 and 17 types of haplotypes identified at MS1, MS3, MS4 and MS7, respectively. These MLGs were distributed among 11(EbpC, EbpA, CHS5, O, KIN-1, PigEBITS5, CHG7, PigEb4, CTS3, GXP-5, GXP-2) of the 22 genotypes identified in this study. The genotype EbpC, forming 13 types of MLGs, was identified as the most diverse genotype, followed by EbpA, which included 11 types of MLGs. The analysis of evolutionary distances based on the tandem sequences of MS1, MS3, MS4, and MS7 showed that 22 genotypes were identified based on ITS. The same genotypes were also separated into different clusters (Fig. 2). These results indicated the complexity of the population structure of E. bieneusi, and suggested that not all individuals with the same ITS genotype belong to a closely related group. Meanwhile, it also demonstrates that these genotypes have undergone complex genetic changes during the evolutionary process. Fig. 2. Molecular phylogenetic analysis of Enterocytozoon bieneusi strain genotypes based on concatenated sequences (MS1, MS3, MS4, and MS7) was performed using the maximum likelihood method. A maximum likelihood phylogenetic tree was constructed using MEGA 11 software (https://www.megasoftware.net) based on 57 Enterocytozoon bieneusi MLG nucleotide sequences, with the Kimura-2 parameter model and 10,000 bootstrap replications
Discussion
The result of the present study showed an overall E. bieneusi prevalence of 24.55% (177/721) from pigs in Guangxi Zhuang Autonomous Region, China. Compared with the prevalence of E. bieneusi in pigs in other regions of China, the infection rates of this study were similar to that in Fujian Province (24.4%) [40], but lower than those reported in Shanxi Province (78.9%) [41], Xinjiang Province (48.6%) [42] and Henan Province (45.5%) [43]. When compared with the E. bieneusi prevalence in pigs reported from other parts of the world, the E. bieneusi prevalence in this study was higher than that reported from Spain (20.6%) [44], Korea (14.2%) [21], and Eastern Slovakia (5%) [45], while lower than that in Brazil (59.3%) [46], USA (31.7%) [27], and Japan (33.3%) [30, 47]. The variation in E. bieneusi prevalence across regions may be influenced by several factors, including sample size, sampling time, host age and feeding mode, etc. Remarkably, Vietnam is adjacent to Guangxi Zhuang Autonomous Region. In Vietnam, the zoonotic genotype E of E. bieneusi was detected in the feces of AIDS patients [48, 49]. The present study identified 22 E. bieneusi genotypes, comprising 12 known genotypes (EbpA, EbpC, CHS5, PigEb4, CHG19, CTS3, PigEBITS5, CHG7, KIN-1, O, Henan-I, BLC17) and 10 novel genotypes (GXP-1 to GXP-10). These findings underscore the necessity for both countries to adopt targeted interventions to mitigate the zoonotic transmission risk of E. bieneusi.
In this study, a significant difference was observed in the infection rates of E. bieneusi among the four regions investigated (χ2 = 65.716, P < 0.05), which could be attributed to differences in feeding management techniques and breeding conditions employed in each region. In this study, the region with the highest prevalence of E. bieneusi was Nanning, with an infection rate of 43.26%. Nanning, the provincial capital of Guangxi Zhuang Autonomous Region, serves as a transportation hub with a high density of population and efficient circulation. However, unlike other areas in Guangxi, Nanning has a relatively small-scale swine industry. This is mainly attributed to its urbanized nature and limited farmland, which restricts large-scale agricultural expansion. In suburban districts, due to their close proximity to urban centers, pig farming predominantly relies on smallholder production systems. These small farms often lack comprehensive biosecurity measures, as many farmers have limited awareness of biosecurity protocols. As a result, the risk of exposure to pathogens increases, and these distinctive factors are likely to contribute to elevated E. bieneusi infection rates. The result underscores the importance of implementing effective surveillance and control measures in regions with higher infection rates, especially in areas with dense human and animal populations, as these environments present an increased risk for pathogen transmission and potential public health threats. The prevalence of E. bieneusi in pigs among different types exhibited significant variation (χ2 = 60.131, P < 0.05). In this study, the highest infection rates of E. bieneusi were observed in nursery pigs among the different types of pigs, which is consistent with previous reports in pigs from southern China [50]. This result suggests a potential correlation with reduced immunity in nursery pigs. There are several factors that may lead to reduced immunity in nursery pigs. Weaning leads to a complex perceived stress in the nursery pigs, such as change in diet. The change in food from breast milk to feed may result in reduced digestibility of nursery pigs [49]. Additionally, maternal antibody levels in nursery pigs may be reduced, resulting in decreased immunity in pigs [41]. The infection rates of E. bieneusi also showed a statistically significant difference between different feeding modes (χ2 = 32.000, P < 0.05). Free-range pigs had a 2.897 times higher risk of E. bieneusi infection compared to intensive pigs (OR = 2.897; 95% CI [1.915–4.382]), which may be due to the more standardized feeding management techniques and better biosafety system in intensive farms [51]. These results showed that intensive feeding mode can reduce the risk of E. bieneusi infection in pigs.
A total of 10 novel genotypes and 12 known genotypes were identified in this study. Single nucleotide polymorphisms (SNPs) were identified by comparing the ITS gene sequences of the 10 novel genotypes (GXP-1 to GXP-10) with GenBank database. Sequence alignment revealed similarities of these sequence were all over 99.47%. However, only 1 to 2 nucleotide site mutations occurred in these novel genotype sequences. The mutation sites were mainly concentrated at 10 different nucleotide positions. Meanwhile, the sequences comparison of the novel genotype showed a high degree of similarity to the sequences of seven genotypes reported previously (EbpC, KIN-1, PigEBITS5, CTS-3, CHG19, PigEB4, and CHG7), respectively. Nucleotide substitution, insertion, and deletion (indel) events are well - established as the primary driving forces behind genomic evolution [52]. Notably, previous studies have indicated that nucleotide substitution displays a neighboring - site preference; namely, the likelihood of a nucleotide mutation and the type of mutated nucleotide are influenced by adjacent nucleotides [52–54]. Significantly, most of the mutated sites among the sequences in this study conformed to this pattern. These results suggested that E. bieneusi is undergoing continuous evolution. In this study, genotypes EbpC and EbpA emerged as the most prevalent ones. Notably, our analyzed results revealed that the identified genotypes GXP-1 and GXP-2 shared a high degree of homology with EbpC. We speculate that the novel genotypes GXP-1 and GXP-2 might be new genotypes that originated from the mutation of EbpC. Moreover, genotypes EbpA and EbpC exhibit an extensive host range, encompassing non-human primates (NHPs), livestock species such as cattle, buffalo, sheep, and goats, domestic pets like dogs and horses, wild animals including deer, foxes, raccoons, bears, pandas, and otters, as well as various avian species such as pigeons, cranes, and parrots [9]. The genotype EbpC and genotype EbpA, with the prevalence of 27.68% and 20.34% respectively, were the dominant genotypes in this study, which are consistent with the results of other studies. These two genotypes have been widely reported in pigs from different regions of China [20], such as Fujian, Jilin, Henan [43], Heilongjiang [14], Zhejiang and Guangdong [55]. This indicates that genotypes EbpC and EbpA are the main prevalent genotypes in pigs in China. The high prevalence of genotypes EbpC and EbpA in China could be attributed to the following factors. The intensive pig farming practices in China create a suitable environment for the transmission and persistence of these genotypes. In such high-density farming settings, pathogens can easily spread from one animal to another [56]. The genotypes O, BLC17 and CTS3 were detected in pigs for the first time in this study, which had previously been reported in dogs [57], bullfrogs [58] and Tibetan sheep [59], respectively. This result demonstrates the potential for cross-species transmission of E. bieneusi. The genotypic identification of E. bieneusi will be further executed in these three species in Guangxi Zhuang Autonomous Region. The results of the phylogenetic analysis showed that all 22 identified genotypes were clustered into zoonotic group 1. The clustering analysis indicated that the novel genotypes identified in this study and the known ones possess zoonotic potential, which highlighting the importance of continuous surveillance and control measures to prevent possible transmission to humans.
MLST analysis has been widely used to the study of population genetics of E. bieneusi [60]. In this study, among 177 E. bieneusi-positive samples, out of 57 samples could be amplified at four loci (MS1, MS3, MS4, and MS7) through multilocus genotyping, yielding 44 different MLGs. There were 19, 10, 19 and 17 types of haplotypes identified at MS1, MS3, MS4 and MS7, respectively. These MLGs were distributed to the identified 11 genotypes (EbpC, EbpA, CHS5, O, KIN-1, PigEBITS5, CHG7, PigEb4, CTS3, GXP-5, GXP-2) in this study. In addition, Multiple MLGs were identified within the same ITS genotype of E. bieneusi, indicating the genetic diversity of E. bieneusi in pigs from Guangxi Zhuang Autonomous Region. In investigations of E. bieneusi in pigs from other parts of China, MLST has also been used in multilocus genotyping of E. bieneusi. Previously reported studies on pigs E. bieneusi multilocus genotyping from Fujian [40], Shaanxi [5] and Sichuan Province [61], 52, 109 and 12 positive samples amplified simultaneously at four loci formed 48, 87 and 10 MLGs, respectively. These studies clarified the high genetic diversity and the complex population structures of E. bieneusi in pigs. However, based on compared MLGs with ITS, a lower number of genotypes were identified by MLGs in present study. Previous study based on MLST analysis have shown that the amplification efficiencies of different loci differ substantially: MS1 has an efficiency of 50.16%, MS3 42.95%, MS4 46.61%, and MS7 41.97% [62]. Furthermore, reported research revealed that the high mutation rate in the E. bieneusi genome can also hinder the effective amplification of specific isolates [63]. Therefore, the development of additional dependable and efficient genetic markers is essential in the future.
Conclusions
This study is the first to report that the infection rates of E. bieneusi in pigs from Guangxi Zhuang Autonomous Region were 24.55% (177/721). Among the different husbandry parameters, pigs living in Nanning, nursery pigs and free-range pigs were more likely to be infected with E. bieneusi. Twelve known genotypes (EbpC, EbpA, PigEb4, Henan-1, PigEBITS5, CHS5, CTS3, CHG7, CHG19, O, KIN-1 and BLC7) and ten novel genotypes (GXP-1 to GXP-10) were identified in this study, and all 22 E. bieneusi genotypes belonged to the zoonotic Group 1. In addition, a total of 44 distinct MLGs were identified by characterizing genetic diversity of E. bieneusi isolates from pigs using the MLST technique. As the first report on E. bieneusi prevalence and genotypes in pigs from Guangxi Zhuang Autonomous Region, this study contributes valuable knowledge to the understanding of the geographical distribution and the genetic diversity of E. bieneusi in pigs.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplementary Material 1: Fig. S1. PCR results of partial samples of ITS, MS1, MS3, MS4, and MS7 genes
Supplementary Material 2
Supplementary Material 3: File S1: Multilocus genotypes of E. bieneusi isolates from pigs
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Stentiford GD, Becnel JJ, Weiss LM, Keeling PJ, Didier ES, Williams BAP, Bjornson S, Kent ML, Freeman MA, Brown MJF, Troemel ER, Roesel K, Sokolova Y, Snowden KF, Solter LF. Microsporidia-Emergent pathogens in the global food chain (Trends in parasitology 32, 336–348; April 2, 2016). Trends Parasitol. 2016;32(8):657.10.1016/j.pt.2016.06.00227365191 · doi ↗ · pubmed ↗
- 2Tavalla M, Kazemi F, Kateki MM, Abdizadeh RJJJM. Molecular diagnosis of Enterocytozoon bieneusi and encephalitozoon spp. Wild Rats Southwest Iran. 2018;11.
- 3WHO. The Food and Agricultural Trade Dataset. [(accessed on 22 April 2025)] Available online. https://www.fao.org/faostat/zh/#country/41