The haplotype-resolved assembly of COL40 a cassava (Manihot esculenta) line with broad-spectrum resistance against viruses causing Cassava brown streak disease unveils a region of highly repeated elements on chromosome 12
Corinna Thoben, Boas Pucker, Stephan Winter, Bethany Fallon Econopouly, Samar Sheat

TL;DR
Researchers assembled the genome of a cassava line resistant to a major viral disease, revealing genetic factors that could help breed more resilient crops.
Contribution
A high-quality, haplotype-resolved genome assembly of a virus-resistant cassava line, COL40, with insights into resistance-linked structural variations.
Findings
COL40 shows broad-spectrum resistance to all known cassava brown streak disease virus strains.
The genome assembly reveals structural variations, including transposable elements and inversions, potentially linked to resistance.
The resource will aid in breeding virus-resistant cassava varieties for improved food security in Africa.
Abstract
Cassava (Manihot esculenta Grantz) is a vital staple crop for millions of people, particularly in Sub-Saharan Africa, where it is a primary source of food and income. However, cassava production is threatened by several viral diseases, including cassava brown streak disease (CBSD), which causes severe damage to the edible storage roots. Current cassava varieties in Africa lack effective resistance to this disease, leading to significant crop losses. We investigated the genetic diversity of cassava and identified new sources of resistance to the viruses causing CBSD. The cassava line, COL40, from a South American germplasm collection showed broad-spectrum resistance against all known strains of the viruses that cause this disease. To further understand the genetic basis of this resistance, we sequenced the genome of COL40 and produced a high-quality, haplotype-resolved genome assembly.…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
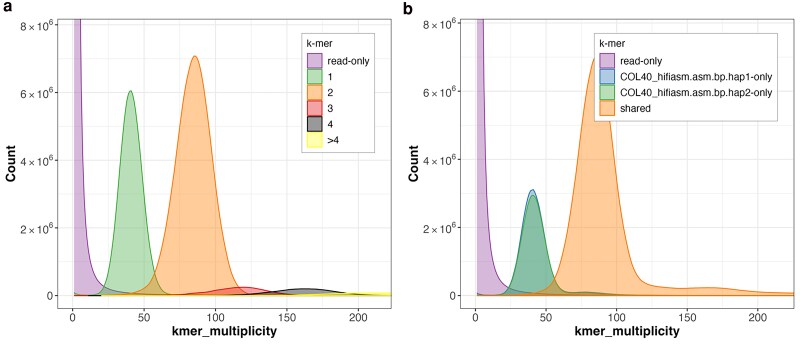 Fig. 1
Fig. 1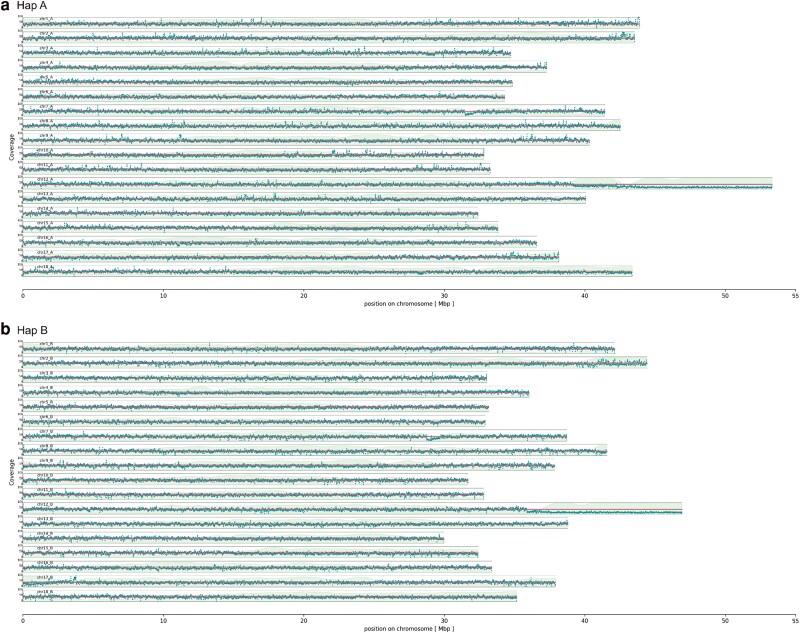 Fig. 2
Fig. 2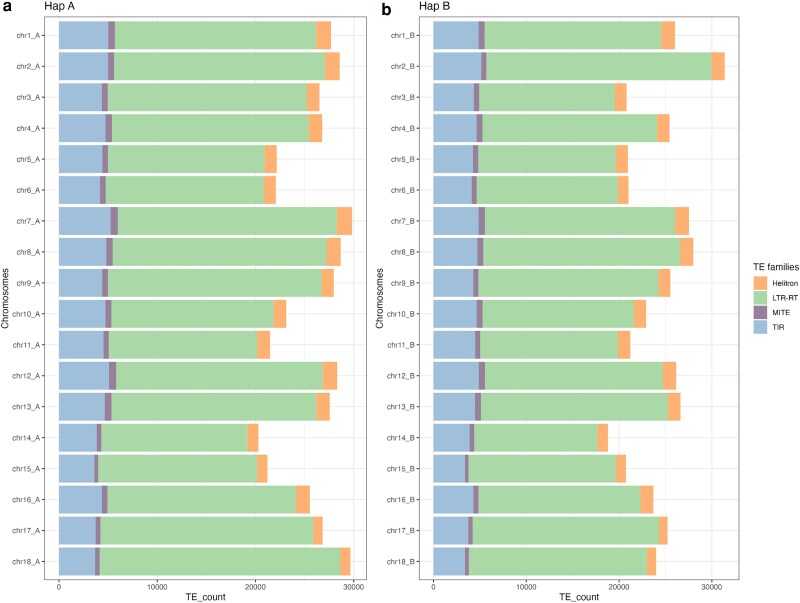 Fig. 3
Fig. 3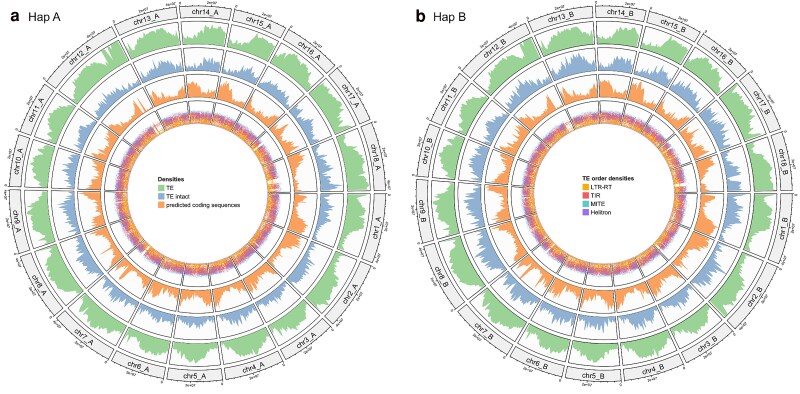 Fig. 4
Fig. 4| Hifiasm | NextDenovo | Flye | Shasta | |
|---|---|---|---|---|
| Assembly size | 823 Mbp, 694 Mbp | 804 Mbp | 693 Mbp | 1,015 Mbp |
| Number of contigs | 2605, 446 | 394 | 9217 | 7692 |
| N50 of contigs | 35 MB, 30 Mbp | 6.7 Mbp | 119 kbp | 1.6 Mbp |
| Number of scaffolds | 2600, 427 | 224 | 6976 | 6334 |
| N50 of scaffolds | 37 MB, 36 Mbp | 41 Mbp | 545 kbp | 41 Mbp |
| BUSCO | 98.8% (D: 8.6%), 98.8% (D: 8.4%) | 99.0% (D: 13.4%) | 98.8% (D: 17.2%) | 75.5% (D: 58.9%) |
| K-mer completeness | 82, 81 (asm: 98) | 86 | 86 | 82 |
| QV | 57, 63 (asm: 58) | 50 | 36 | 61 |
| COL40 | TMEB117 | TME204 | TME3 | |
|---|---|---|---|---|
| Chromosomes resolved | yes | yes | yes | No |
| Haplotype-resolved | yes | yes | yes | No |
| Assembly size | 823 MB, 694 Mbp | 694 MB, 665 Mbp | 762 MB, 706 Mbp | 1,225 Mbp |
| N50 of scaffolds | 37 MB, 36 Mbp | 38 MB, 36 Mbp | 18 MB, 26 Mbp | 53 Mbp |
| BUSCO | 98.8% (D: 8.6%), 98.8% (D: 8.4%) | 98.9% (D: 8.8%), 98.9% (D: 8.8%) | 99% (D: 4.9%), 98.8% (D: 4.2%) | 94.8% (D: 19.7%) |
| K-mer completeness | 82%, 81% | 78.63%, 77.95% (asm: 98.79%) | 79.6%, 79.1% | − |
| QV | 57, 63 | 64, 68 | 45, 49 | − |
| BUSCO (Predicted polypeptide sequences) | 96.6% (D: 22.7%), | ∼90% both | 96.7% (D: 17.4%), 96.7% (D: 16%) | − |
| TE proportion | 58.04% | 57.37%, 54.42% | >60% | 64.81% |
| Reference | This study |
|
|
|
- —Bill & Melinda Gates Foundation10.13039/100000865
- —Next Generation Cassava Breeding
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsCassava research and cyanide · Nematode management and characterization studies · Genetic and Environmental Crop Studies
Introduction
Cassava (Manihot esculenta) is an important world food crop that produces starch-rich storage, which serves as a main source of food and income for people in Africa, South America, India, and Southeast Asia. The plant originated in the Amazon region of South America but is especially important in Sub-Saharan Africa, where half of the world's cassava is produced mainly by smallholder farmers. This resilient plant adapts well to diverse environments, is resistant to drought, grows in poor soils, and guarantees food security even in the most adverse environmental conditions. Nevertheless, cassava cultivation is constrained by pests and diseases, and because stem cuttings are taken to propagate the crop, diseases are maintained and disseminated by vegetative propagules. Viruses are associated with cassava cultivation in all cassava regions of the world and present serious threats to crop production. In Africa, viruses causing cassava mosaic disease are endemic, but the deployment of varieties with high virus resistance has mitigated the impact of the diseases. In contrast, there was no means to control the spread of the viruses causing cassava brown streak disease (CBSD), which has a severe impact because of the root necrosis destroying the edible tubers of the crop. The relatively recent outbreak, first noted in 2004/2005 in Uganda (Alicai et al. 2007), the limited knowledge about the etiology and epidemiology of the disease, and the lack of virus-resistant cassava in Africa delayed comprehensive actions to control the disease. Cassava brown streak virus (CBSV, species Ipomovirus brunusmanihotis) and Ugandan cassava brown streak virus (UCBSV, species Ipomovirus manihotis) are closely related members of the genus Ipomovirus within the family Potyviridae. Both viruses are the causal agents of CBSD, sharing a high degree of genetic similarity and inducing similar symptoms in infected plants. However, they differ in genome sequences, virulence, and geographical distribution. Notably, CBSV is often associated with more severe symptoms compared with UCBSV, contributing to greater yield losses in affected regions. Searching for new sources of resistance, we explored the diversity of South American cassava. We infected a subset of a germplasm collection held at International Center for Tropical Agriculture with diverse cassava brown streak viruses to identify lines that showed resistance to either isolates of the CBSV or against the UCBSV After stringent infection and virus screening, 3 germplasm accessions COL2182, PER556, and COL40 showed broad-spectrum immunity to all species and strains of the viruses causing CBSD (Sheat et al. 2019). COL40, the most promising line, is now used as a source to provide CBSD resistance essentially in all cassava improvement programs. First prototypes from crosses with African lines are in the field, proving that high virus resistance in progenies from COL40 can be reached. Progress has been made in characterizing the resistance phenotype of COL40 (Sheat et al. 2021) and advancing field evaluation and resistance assessment (Sheat and Winter 2023). However, important steps toward molecular breeding, genomic prediction, marker-assisted selection, and a mechanistic explanation of the virus resistance in COL40 remain open challenges.
The reconstruction of complete, haplotype-resolved genome sequences of cassava genotypes is a substantial achievement in providing genome information and resources to advance our understanding of the cassava genome(s) and accelerate breeding to future-proof the crop for resilience to biotic and abiotic stresses, agronomic traits, and consumer demands. As a highly heterozygous species (Prochnik et al. 2012) (1% in TME 204; Qi et al. 2022) its genome comprises extensive amounts of repetitive elements (TMEB117 >60%; Landi et al. 2023), allelic variation, inversions, and deletions/insertions comprising large segments of the genome (Ramu et al. 2017). This genomic structural variation limits the use of a single reference genome sequence (AM560-2; Phytozome 13, v 8.1) that does not capture the full complement of sequence diversity of a crop species (Jayakodi et al. 2020) and may lack genes present in other genotypes, e.g. NBS-LRR genes relevant for disease resistance (Lozano et al. 2015).
Recently, high-quality haplotype-resolved genome sequences of African cassava lines were reported (Qi et al. 2022; Kuon et al. 2019; Mansfeld et al. 2021; Landi et al. 2023). The lines chosen were TME3 and 60444, to contrast a landrace with high resistance against viruses causing CMD, carrying the CMD2 resistance locus (TME3) and a highly susceptible line 60,444. Similarly, TME204 (Qi et al. 2022), carrying CMD2, and TMEB117 lacking (Landi et al. 2023) were chosen to provide high-resolution genome data for virus studies on resistance mechanisms and, to guide future breeding. Our study follows a similar rationale. By generating a high-quality COL40 genome sequence, we have established a reference genome, to advance our understanding about virus resistance/immunity in cassava. This resource enables the identification of genetic variants associated with resistance traits through genome-wide association studies and supports cassava resistance breeding efforts, with COL40 serving as a valuable genetic source.
Materials and methods
Plant cultivation
Cassava COL40 plantlets grown in tissue culture were transferred to pots and maintained at the greenhouse facility of the DSMZ Plant Virus Department in Braunschweig Germany at temperatures between 26 and 32°C and >75% relative humidity with additional light provided during German winter conditions.
DNA extraction, library preparation and sequencing
Prior to sampling, the plants were transferred to the dark for 24 h, after which fully-expanded leaves (young leaves; between 3rd and 5th leaf) were taken and flash frozen in liquid N_2_. Leaves (10–15 g) were sent to Arizona Genomics Institute for high-molecular-weight (HMW) genomic DNA (gDNA) extraction, library preparation, and sequencing. HMW gDNA extraction was done essentially following the CTAB method of Doyle and Doyle, 1987 (Doyle and Doyle 1987), with minor modifications (Fu et al. 2017). The HiFi express template prep kit 2.0 was used for PacBio HiFi SMRTbell library construction. Fragments between 10 and 25-kb were selected for HiFi sequencing on the Sequel II instrument. SeqII v.2.0 chemistry was used with 2 SMRT cells 8M v1 in circular consensus sequencing mode for a 30-h run time per cell.
For RNA-seq analysis, leaf, stem, and root samples were taken from COL40 and flash frozen in liquid N_2_ prior to extraction of total RNA using a RNA extraction kit following the manufacturer's protocol (Epoch, United States). RNA was quantified in a QubitR fluorometer (Thermo Fisher Scientific, United States) using the Qubit RNA BR Assay Kit (Thermo Fisher Scientific, United States), and checked for size distribution in a bioanalyzer (Agilent) prior to sequencing at Novogene (Science Park, Milton, Cambridge, UK). At Novogene, mRNA was purified from total RNA preparations using poly-T magnetic beads, fragmented and subjected to first strand cDNA synthesis using random hexamer primers followed by second strand cDNA synthesis and end repair. Library construction, quality controls, and paired-end sequencing (PE 150) on an Illumina Novaseq 6000 platform followed Novogene's mRNA sequencing workflow.
Assembly generation and quality control
The HiFi reads of the COL40 cultivar were assembled by Hifiasm 0.19.8-r603 (Cheng et al. 2021, 2022), NextDenovo 2.5.0 (Hu et al. 2024), Flye 2.9.3-b1797 (Kolmogorov et al. 2019), and Shasta 0.11.1 (Shafin et al. 2020). To avoid compatibility issues with tools in downstream analysis, contig identifiers of the assemblies were cleaned using the python script clean_genomic_fasta.py v0.15 (Meckoni, et al 2023). Assembly statistics were calculated with the python script contig_stats.py v1.31 (Pucker et al. 2016). Completeness of universal single-copy orthologs was checked with BUSCO 5.2.2 and the eudicots_odb10 dataset (Manni et al. 2021). A k-mer based analysis was performed with Merqury (Rhie et al. 2020) and k-mers generated from the cultivars HiFi reads (k = 21). The assembly's coverage was analyzed by mapping the HiFi reads to the assembly with minimap2 2.24-r1122 and “-ax map-hifi –secondary = no” option (Li, 2018). Samtools 1.17-29-gcc18465 (Bonfield et al. 2021) was applied for conversion and sorting of the mapping file. The coverage was calculated with the “genomecov” command of bedtools v2.30.0 (Quinlan and Hall 2010).
Scaffolding of the assembly
Genetic markers of the composite genetic map of M. esculenta Crantz by the International Cassava Genetic Map Consortium (ICGMC) (Supplementary File 2, International Cassava Genetic Map Consortium (ICGMC), 2014) were used for scaffolding. The genetic markers were mapped to the assemblies using the python script genetic_map_to_fasta.py v0.2 (https://github.com/c-thoben/CassavaGenomicsProject), which creates a csv map based on the best BLAST hits for each marker with a minimum similarity of 99.0% and score of 175. The scaffolding was performed by converting the CSV map with ALLMAPS (JCVI utility libraries 1.4.2) (Tang et al. 2015) “merge” option and path construction using the “path” option. Scaffold statistics were calculated with the python script contig_stats.py v1.31 (Pucker et al. 2016). For the Hifiasm assembly, the scaffolding was performed for each haplophase separately. The scaffolds of both haplophases were merged together, and for each scaffold, a suffix was added to the scaffold IDs indicating its haplophase. The coverage plot of the chromosomes including the density of the transposable elements was created with the python script chromosome_coverage_te_plot.py v0.4 (Pucker et al. 2016; https://github.com/c-thoben/CassavaGenomicsProject).
Prediction and functional annotation of polypeptide sequences
In total, 21 RNA-Seq libraries of the COL40 cultivar (Supplementary Additional File 5) and 24 further RNA-Seq libraries (Supplementary Additional File 5) from other cultivars were mapped to the COL40 assembly with HISAT2 2.2.1 (Kim et al. 2019) and “–dta” option. Samtools 1.17-29-gcc18465 (Danecek et al. 2021) was applied for conversion and sorting of the mapping files (Bonfield et al. 2021). Together with external protein hints from M. esculenta Crantz v8.1 (International Cassava Genetic Map Consortium (ICGMC), 2014) and the OrthoDB 11 Viridiplantae database (Kuznetsov et al. 2023), the mapped RNA-Seq reads were given to BRAKER3v3.0.6 (Stanke et al. 2006, 2008; Gabriel et al. 2021, 2024) as external hints to perform the structural annotation. Based on the mapped RNA-Seq reads, the coverage of the structural annotation was analyzed with the python script RNAseq_cov_analysis.py v0.1 (Meckoni et al. 2023; https://github.com/bpucker/GenomeAssembly/) and filtered for transcripts with a coverage >90%. Completeness of the predicted polypeptide sequences was checked with BUSCO v5.2.2 and the eudicots_odb10 (Manni et al. 2021) dataset for each haplophase separately. The polypeptide sequences were functionally annotated using InterProScan5 v5.67-99.0 (Manni et al. 2021; Blum et al. 2021).
Prediction and analysis of transposable elements
Transposable elements were predicted with EDTA v2.1.0 (Ou et al. 2019) and the “–overwrite 1 –sensitive 1 -anno 1 -evaluate 1” options. Annotated coding sequences predicted by BRAKER3 were provided with the “–cds” option. Annotated transposable elements were further analyzed with the script COL40_TE_repeat_analysis.ipynb (https://github.com/c-thoben/CassavaGenomicsProject) based on the R script TMEB117TEandGeneAnnotation.R provided by (Landi et al. 2023; https://github.com/LandiMi2/GenomeAssemblyTMEB117). The R package circlize (Gu et al. 2014) was used to calculate the density of transposable elements and predicted coding sequences with the circos function genomicDensity and visualize them in a circos plot. The coverage plot of the chromosomes including the mean coverage over 1 kbp and the density of the transposable elements over 1 Mbp was created with the python script coverage_te_plot.py v0.5 (Pucker et al. 2016; https://github.com/c-thoben/CassavaGenomicsProject).
Results
Assembler comparison and quality control
The HiFi dataset consisted of 3,440,114 reads with an N50 length of 18,106 and 38% GC content. All assemblies were >80% complete representations of the genome according to the k-mer analysis and, with exception of the Shasta assembly, show a high completeness regarding the single-copy universal orthologs. The Hifiasm (N50: 35 Mbp, 30 Mbp) and NextDenovo (N50: 6.7 Mbp) assemblers performed best in producing assemblies with a high continuity (Table 1).
The Hifiasm assembly had the highest k-mer completeness (98%, ∼80% for each haplophase) and a good assembly consensus quality value (QV) of 58 according to the k-mer analysis. The analysis of the k-mer copy numbers (Fig. 1) demonstrates that the haplotypes are well-resolved in this assembly.
Assembly completeness according to k-mer analysis of the assembly. a) Merqury copy number spectrum plot of the HiFi reads. The green peak represents 1-copy reads. The orange peak represents 2-copy reads. HiFi reads not included in the assembly are represented by the purple peak. b) Copy number spectrum plot of the k-mers in A. Heterozygous reads are colored green or blue according to the haplophase, overlapping peaks. Homozygous reads or haplotype-specific duplications are colored orange.
The Shasta assembly had the highest assembly quality (QV 61) in the k-mer analysis, standing out in this specific criterion, but was outperformed by the Hifiasm assembly in the remaining criteria. Therefore, the Hifiasm assembly was chosen as the representative genome sequence of COL40.
Scaffolding and structural annotation
In the scaffolding process, 16,233 out of 22,403 markers were mapped to the contigs of haplophase A and used to scaffold 23 contigs representing the 18 M. esculenta chromosomes (Supplementary Additional Files 1 and 2). The 18 chromosomes of haplophase B were scaffolded from 37 contigs with reference to 16,169 mapped markers. The remaining 2,582 contigs for haplophase A and 409 contigs for haplophase B remained unplaced. The structural annotation of the Hifiasm assembly predicted 82,151 polypeptide sequences for haplophase A and 72,383 polypeptide sequences for haplophase B. After filtering the transcripts based on their coverage in the mapped RNA-Seq reads, the number of sequences was reduced to 36,064 for haplophase A and 34,029 for haplophase B. The BUSCO analysis revealed a high completeness of 96.6% (D: 22.7%) for haplophase A, 96.7% (D: 21.7%) for haplophase B, and 98.6% (D: 94.6%) for both haplophases.
Assembly coverage analysis
The coverage analysis revealed an average coverage of 40.4-fold for haplophase A and 39.7-fold for haplophase B. The coverage plot demonstrates an equal distribution of the coverage, which can be attributed to the high-resolution of the haplophases in the assembly (Supplementary Additional Files 3 and 4). For both haplophases, a coverage drop at the end of chromosome 12 (downstream of ∼ 39 Mbp for haplophase A, ∼ 37 Mbp for haplophase B) is displayed in the coverage plot (Fig. 2). This is also reflected in the coverage histogram of chromosome 12, which shows a second peak around a coverage of 15 for both haplophases (Supplementary Additional Files 3 and 4). For both haplophases, the density of transposable elements is enhanced in these regions, which is further discussed below (Figs. 2 and 3).
Coverage plot of the chromosome-resolved Hifiasm assembly. The HiFi reads were mapped to the haplophase A a) and haplophase B b) of the scaffolded assembly and the mean coverage was calculated for each haplophase (red line) and block-wise over 1 kbp intervals for each chromosome (dark green dots). The density of transposable elements was calculated block-wise per1 Mbp interval (light green area).
Distribution of transposable element families for pseudochromosomes of haplophase A a) and haplophase B b) in the Hifiasm assembly. Unclassified transposable elements were removed from the annotation. For all pseudochromosomes, LTR-RTs are the most abundant repeats for both haplophases.
Annotation of transposable elements
In total, 3,027 repeat regions were annotated, which cover 58.04% of the COL40 assembly. The most abundant transposable elements (TL) are long terminal repeats—retrotransposons (LTR-RTs), mostly Gypsy LTR-RT, which cover 51.88% of the complete assembly and are the most abundant family on all chromosomes (Fig. 3).
As described above, the density of TE increases at the end of chromosome 12 in both haplophases (Fig. 4). This increase can be attributed to fragmented transposable elements, as the density of structurally intact transposable elements decreases. Furthermore, the density of predicted coding sequences is distinctly reduced in this region. The scaffolding results (Supplementary Additional Files 1 and 2) show that no markers of the M. esculenta Crantz genetic map by the International Cassava Genetic Map Consortium (ICGMC) [Supplementary File 2, International Cassava Genetic Map Consortium (ICGMC), 2014] were mapped to the region, suggesting that the transposable elements in this area are either not represented in the map or that markers could not be developed due to the high level of repetitive sequences present in the region.
Density of transposable elements and predicted coding sequences across the pseudochromosomes of haplophase A a) and haplophase B b) in the Hifiasm assembly. For all tracks, the density was calculated over a window size of 1 Mbp. The density of transposable elements is shown in the outer track and reflects the percent of the window covered by the input regions (green). The second track shows the density of structurally intact transposable elements (blue). The density of predicted coding sequences is displayed in the third track (orange). In the inner track, a rainbow plot represents the minimal distance (log10-transformed) to the neighboring regions. The color of the region displays the transposable elements order.
Discussion
In this study, we presented a haplotype-resolved genome assembly of the cassava line COL40, a South American germplasm line with high and broad-spectrum resistance to the viruses causing CBSD. The assembly of COL40 joins a growing collection of high-quality genome assemblies from other cassava cultivars, such as TMEB117, TME204, and TME3 (Kuon et al. 2019; Qi et al. 2022; Landi et al. 2023), and highlighting the increasing role of advanced sequencing technologies in improving crop genomics.
Using PacBio HiFi reads and after a comparative evaluation of assembly tools, we selected Hifiasm as the optimal assembler due to its superior continuity and completeness. Hifiasm also generated best assembly in other cassava genome studies (Kuon et al. 2019; Landi et al. 2023) with haplophase sizes between 665 and 823 Mbp (Table 2).
The high accuracy of HiFi data was crucial for achieving a robust genome assembly with phased haplotypes. Similarly, PacBio HiFi sequencing reported for the assembly of other plant genomes, including Haloxylon ammodendron (Wang et al. 2022), and Rhododendron vialii (Chang et al. 2023) allowed generation of highly contiguous genome assemblies for diploid and polyploid species and made it ideal for resolving the complex structure of cassava's genome.
However, we recognize that Oxford Nanopore Technology has made significant advancements, particularly in read length and base-calling accuracy, allowing for highly contiguous and scaffolded genome assemblies. As sequencing technologies continue to evolve, leveraging the strengths of both approaches may offer the most comprehensive and accurate genome reconstructions.
Assemblies comparison among published data and our data present a high completeness of >98% in both BUSCO and k-mer analysis and around 80% for each haplophase in k-mer analysis (Table 2).
The observed duplication of BUSCOs among the predicted proteins is a result of the assembly being derived from the merging of haplophase A and haplophase B. Since most regions of the assembly are represented in both haplophases, the assembly predominantly exhibits a diploid nature, leading to duplication. Additionally, regions that were already duplicated within one or both haplophases before merging naturally appear as triploid or tetraploid in the final assembly. This explains the higher number of duplicate BUSCOs detected in the predicted proteins compared with those directly identified on the scaffolds.
Our results are consistent with other assemblies of complex plant genomes like that of the European common bean (Carrère et al. 2023). This level of completeness indicates that our assembly has captured the vast majority of the gene space, an essential feature for downstream functional studies, particularly for understanding CBSD resistance mechanisms.
Comparing the QV of different cassava assemblies (Table 2), we observed that the COL40 genome assembly had a QV of 57 for one haplotype and 63 for the other, which is close to the TMEB117 assembly (Landi et al. 2023). In contrast, earlier assemblies, such as TME204 (Qi et al. 2022), had significantly lower QVs. These improvements in genome quality metrics reflect the advancements made by HiFi sequencing, as highlighted by Sun et al., (2022), where HiFi reads produced near-perfect assemblies for various complex plant genomes.
Another outstanding feature of our assembly is that we resolved high-quality polypeptide sequences, with a completeness of around 97% for both haplotypes. These results align with other high-quality plant genome assemblies produced using HiFi data, such as the assembly of Salix wilsonii (Han et al. 2022) and the bean genome (Carrère et al. 2023). These findings underscore the value of using HiFi reads, as they produce long, highly accurate reads that are crucial for resolving complex regions and providing reliable gene annotations.
The COL 40 assembly provides critical insights into the repetitive regions and structural variations in cassava genomes. The coverage analysis along the pseudochromosomes after scaffolding of the COL40 assembly revealed a region with reduced coverage at the end of chromosome 12 on both haplophases (Fig. 2). The annotation of this assembly region showed a high density of fragmented transposable elements TE (Fig. 4), which was also found in chromosome 12 of TMEB117 (Landi et al. 2023). This region is not covered by genetic markers in the composite genetic map of M. esculenta Crantz by the International Cassava Genetic Map Consortium (ICGMC) (Supplementary Additional File 2), suggesting that the TEs in this region are either not represented or, that the high frequency of repetitive sequences hindered the developed of respective markers. Nevertheless, this region may not be present in all cassava genomes but it is also likely that earlier assemblies based on short sequencing reads may have not resolved repetitive regions and consequently created gaps and fragmentation of the transposable elements. Highly TE-rich regions present challenges to the completeness of genome assemblies, for cassava and other species (Benham et al. 2024).
The COL40 assembly focuses on haplotype resolution and genome completeness. In contrast, the telomere-to-telomere assembly of the cassava Xinxuan 048 delved deeper into epigenetic analysis (Xu et al. 2023), highlighting that the structural variation between the haploid genomes of Xinxuan 048 was mainly due to TE insertions. The variation led to differences in CG methylation of alleles, thus resulting in differential allelic expression. These findings suggest that differential gene expression in Xinxuan 048 could be influenced by methylation changes caused by varying TE insertions.
The proportion of TE elements in the genome of COL 40 is around 60%, and this is consistently found in all cassava genome assemblies (Kuon et al. 2019; Landi et al. 2023). These TE-rich regions may hold essential genes for interesting traits, such as those associated with CBSD resistance.
In conclusion, the high-quality, haplotype-resolved genome assembly of cassava COL40 contributes significantly to cassava genomic resources. It lays the groundwork for further comparative analyses between cassava cultivars and provides an invaluable resource to advance our understanding of virus resistance and immunity.
Supplementary Material
jkaf083_Supplementary_Data
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Alicai T, Omongo C, Maruthi M, Hillocks R, Baguma Y, Kawuki R, Bua A, Otim-Nape G, Colvin J. 2007. Re-emergence of cassava brown streak disease in Uganda. Plant Dis. 91(1):24–29. doi:10.1094/PD-91-0024.30781061 · doi ↗ · pubmed ↗
- 2Benham PM, Cicero C, Escalona M, Beraut E, Fairbairn C, Marimuthu MPA, Nguyen O, Sahasrabudhe R, King BL, Thomas WK, et al 2024. Remarkably high repeat content in the genomes of sparrows: the importance of genome assembly completeness for transposable element discovery. Genome Biol Evol. 16(4):evae 067. doi:10.1093/gbe/evae 067.38566597 PMC 11088854 · doi ↗ · pubmed ↗
- 3Blum M, Chang H-Y, Chuguransky S, Grego T, Kandasaamy S, Mitchell A, Nuka G, Paysan-Lafosse T, Qureshi M, Raj S. 2021. The Inter Pro protein families and domains database: 20 years on. Nucleic Acids Res. 49(D 1):D 344–D 354. doi:10.1093/nar/gkaa 977.33156333 PMC 7778928 · doi ↗ · pubmed ↗
- 4Bonfield JK, Marshall J, Danecek P, Li H, Ohan V, Whitwham A, Keane T, Davies RM. 2021. HT Slib: C library for reading/writing high-throughput sequencing data. Gigascience. 10(2):giab 007. doi:10.1093/gigascience/giab 007.33594436 PMC 7931820 · doi ↗ · pubmed ↗
- 5Carrère S, Mayjonade B, Lalanne D, Gaillard S, Verdier J, Chen NW. 2023. First whole genome assembly and annotation of a European common bean cultivar using Pac Bio Hi Fi and iso-seq data. Data Brief. 48:109182. doi:10.1016/j.dib.2023.109182.37383758 PMC 10293967 · doi ↗ · pubmed ↗
- 6Chang Y, Zhang R, Ma Y, Sun W. 2023. A haplotype-resolved genome assembly of Rhododendron vialii based on Pac Bio Hi Fi reads and Hi-C data. Sci Data. 10(1):451. doi:10.1038/s 41597-023-02362-1.37438373 PMC 10338486 · doi ↗ · pubmed ↗
- 7Cheng H, Concepcion GT, Feng X, Zhang H, Li H. 2021. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat Methods. 18(2):170–175. doi:10.1038/s 41592-020-01056-5.33526886 PMC 7961889 · doi ↗ · pubmed ↗
- 8Cheng H, Jarvis ED, Fedrigo O, Koepfli K-P, Urban L, Gemmell NJ, Li H. 2022. Haplotype-resolved assembly of diploid genomes without parental data. Nat Biotechnol. 40(9):1332–1335. doi:10.1038/s 41587-022-01261-x.35332338 PMC 9464699 · doi ↗ · pubmed ↗