Proteinuria in Alport Syndrome: Treatment With Sodium-Glucose Co-Transporter-2 (SGLT2) Inhibitors
Gautam Agrawal, Bhawna Agarwal, Pallavi Shirsat, Kunal Sonavane

TL;DR
This case report discusses the treatment of a woman with Alport syndrome using SGLT2 inhibitors to manage kidney disease.
Contribution
The paper highlights the potential use of SGLT2 inhibitors in managing Alport syndrome, a genetic disorder.
Findings
A 40-year-old female with Alport syndrome was managed using ACE inhibitors and SGLT2i to prevent kidney damage.
Genetic testing and multidisciplinary care are emphasized for effective treatment of Alport syndrome.
Limited research exists on the benefits of SGLT2 inhibitors in genetic disorders like Alport syndrome.
Abstract
Alport syndrome (AS) is a genetic disorder characterized by progressive kidney disease, hearing loss, and eye abnormalities. It is caused by mutations in the genes responsible for producing type IV collagen, which is a crucial component of the glomerular basement membrane, the cochlea, and the lens of the eye. We present a case of a 40-year-old female who presented with persistent microscopic hematuria and proteinuria and was diagnosed with autosomal dominant AS based on kidney biopsy and genetic testing. This case report discusses the clinical presentation, diagnostic work-up, and management approach of patients with AS. We highlight the current advancement in management of CKD with SGLT 2 inhibitors, with limited research regarding the benefit of sodium-glucose cotransporter-2 inhibitors (SGLT2i) in patients with genetic disorders like AS. Early recognition and management of AS with…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
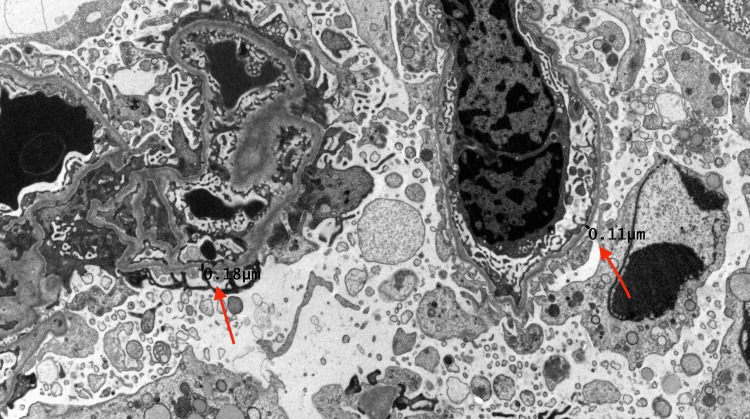 Figure 1
Figure 1| Laboratory tests | Results | Reference range |
| 24-hour protein | 2141 | < 150 mgs |
| Serum albumin | 3.8 | 3.5-5.7 gm/dl |
| Blood urea nitrogen (BUN) | 9 | 7-25 mg/dl |
| Creatinine | 0.7 | 0.6-1.2 mg/dl |
| Calcium | 9 | 8.6 - 10.3 mg/dl |
| Hemoglobin | 13 | 11.7-15.8 gm/dl |
| ANA | 0.42 | <0.8-Negative |
| Anti-ds DNA | Negative | <25-Negative |
| Hemoglobin A1c | 5.1 | 5.7-6.4 (Prediabetic) % |
| Hepatitis B surface antigen | Non-reactive | Non-reactive |
| Hepatitis C antibody | Non-reactive | Non-reactive |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsDiabetes Treatment and Management · Cell Adhesion Molecules Research · Platelet Disorders and Treatments
Introduction
Alport syndrome (AS), also known as hereditary nephritis, is a hereditary disorder characterized by progressive renal disease, sensorineural hearing loss, and ocular abnormalities. It is caused by mutations in the COL4A3-5 genes, which encode the alpha3-5 chains of type IV collagen, a crucial component of the glomerular basement membrane (GBM) in kidneys, the cochlea in the ear and the retina in the eye. The mode of inheritance can be X-linked, autosomal dominant, autosomal recessive, or rarely digenic inheritance. AS is the most common cause of inherited kidney disease and the second most common genetic cause of kidney failure [1,2]. Renal manifestations include hematuria, proteinuria, hypertension, and progression to kidney failure. Diagnosis is confirmed by molecular genetic testing or skin or kidney biopsy.
Despite the increasing availability of genetic testing, tissue studies remain essential for evaluating Alport syndrome (AS) in patients with persistent hematuria. Kidney biopsy findings often reveal thinning of the glomerular basement membrane [3]. Currently, there is no specific cure for AS, and patients who develop renal failure typically require kidney transplantation. The standard of care to slow the progression of renal deterioration involves treatment with angiotensin-converting-enzyme (ACE) inhibitors [3]. Initiating therapy at a higher glomerular filtration rate (GFR) has been shown to delay the onset of kidney failure. Sodium-glucose cotransporter 2 (SGLT2) inhibitors can reduce albuminuria and slow the progression of chronic kidney disease [4], although data regarding their efficacy in patients with AS remains limited.
This case report discusses the clinical presentation, diagnostic workup, and management of AS in a 40-year-old female patient. It highlights ongoing research in interventions to slow renal disease progression and the need for further research regarding the use of sodium-glucose cotransporter-2 inhibitors (SGLT2i) in patients with AS.
Case presentation
This is the case of a 40-year-old female who had pertinent past medical history of a solitary episode of kidney stone (which she passed spontaneously), irritable bowel syndrome, depression, and a ruptured ovarian cyst. She denied any known family history of hereditary kidney disease.
She was found to have persistent proteinuria and microscopic hematuria, for which she was seen in the nephrology clinic. On physical examination, her weight was 210 lbs, and her blood pressure was 120/78 mm Hg. She appeared euvolemic on examination, with no peripheral edema. Her lungs were clear to auscultation bilaterally, and her heart sounds were regular. She had no rash, lymphadenopathy, oral ulcers, or tattoos. As shown in Table 1, her laboratory values demonstrated sub-nephrotic proteinuria, with a 24-hour protein level of 2,141 milligrams. She had normal renal function, with a serum creatinine of 0.7 mg/dL. Serological workup showed a negative anti-nuclear antibody, negative anti-double-stranded DNA antibody, normal complement levels, and a serum protein electrophoresis that was negative for a monoclonal spike. Her hemoglobin A1C was 5.1%.
She was referred for a kidney biopsy, which revealed diffuse thinning of the glomerular basement membrane as shown in Figure 1, with a mean thickness of 178 nanometers (normal range of 250-330 nanometers), raising the possibility of Alport syndrome.
Kidney biopsy slide showing thinning of glomerular basement membraneRed arrow indicating glomerular basement membrane measurements.
Genetic testing showed a monoallelic mutation in the COL4A4 gene, which was likely pathogenic and consistent with an autosomal dominant inheritance pattern. She was started on lisinopril, which was gradually up-titrated to the maximum tolerated dose. Her proteinuria improved to 1246 mg in the 24-hour urine test. She was then started on dapagliflozin for renal protection. She has tolerated dapagliflozin well, with no episodes of urinary tract infections, hypotension, or dehydration. Her proteinuria has fluctuated but improved overall, with the 24-hour protein level of 1007 mg after one year of therapy. Her renal function has remained stable, with a serum creatinine of 0.7 mg/dL nearly three years post-treatment.
Discussion
This case underscores the importance of considering AS in patients with unexplained hematuria or proteinuria, particularly when there is a family history of similar symptoms. Early diagnosis and comprehensive management are essential to slow the progression of kidney disease and prevent complications. AS is a genetically and phenotypically heterogeneous disorder. Clinical presentations can be variable depending on the mode of inheritance or genetic variant. X-linked Alport syndrome commonly results in end-stage renal disease (ESRD) in males, affecting approximately 50% by age 25 and 90% by age 40 [2]. Over 90% of patients with autosomal recessive AS progress to end-stage kidney disease (ESKD) by the age of 40 [2]. Patients with autosomal dominant usually have a slower progression to ESRD.
Early intervention with ACE inhibitors or angiotensin receptor blockers (ARBs) can reduce proteinuria and delay the progression of renal disease [3], as noticed in our patient. SGLT2i reduces albuminuria independently of glucose control by lowering tubular glucose reabsorption, blood pressure, and intraglomerular pressure, key factors in slowing chronic kidney disease (CKD) progression [4]. However, the efficacy of SGLT2i in underrepresented subgroups, as patients with AS or immune-mediated glomerular disorders, remains uncertain, as they may affect lean individuals whose CKD progression may involve alternative pathways like inflammation or genetic abnormalities [5]. Some evidence suggests that the renal benefits of SGLT2i may be influenced by body mass index (BMI), with greater effects observed in patients with higher BMI [5]. Given these considerations, dedicated clinical trials are necessary to assess the efficacy of SGLT2i in patients with this patient population.
A case series involving patients with hereditary podocytopathies, including Alport syndrome, reported that treatment with SGLT2i significantly reduced the urinary albumin-creatinine ratio (UACR), indicating a potential benefit in reducing proteinuria and slowing renal dysfunction [6]. However, due to the limited treatment duration and small sample size, more robust studies are needed to validate these results. There is an ongoing trial named the DOUBLE PRO-TECT Alport Trial, which is a multicenter, randomized, double-blind, placebo-controlled trial that aims to assess the safety and efficacy of dapagliflozin, an SGLT2i, in young patients with Alport syndrome. The primary outcome is the change in UACR from baseline to week 48, with secondary outcomes including changes in estimated glomerular filtration rate (eGFR) [7]. Renal transplantation is the definitive treatment for ESRD. Given the genetic nature of the disease, counseling for the patient and their family is essential.
Conclusions
Early intervention with ACE inhibitors or angiotensin receptor blockers (ARBs) is recommended to reduce proteinuria and slow the progression of renal disease in Alport syndrome. However, the efficacy of SGLT2 inhibitors (SGLT2i) in underrepresented subgroups, such as patients with Alport syndrome, remains uncertain. This case highlights the importance of early recognition and diagnosis of Alport syndrome to enable timely therapeutic intervention with dual therapy using ACE inhibitors and SGLT2i, aiming to slow disease progression and prevent complications. Further research and long-term data are needed to better understand the use of SGLT2i in patients with Alport syndrome.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Alport syndrome: an update Curr Opin Nephrol Hypertens Savige J 206211342025 https://doi.org/10.1097/MNH.00000000000010633984058910.1097/MNH.0000000000001063 · doi ↗ · pubmed ↗
- 2Alport syndrome Adv Kidney Dis Health Chavez E Goncalves S Rheault MN Fornoni A 170179312024 https://doi.org/10.1053/j.akdh.2024.02.0043900445710.1053/j.akdh.2024.02.004 · doi ↗ · pubmed ↗
- 3Alport syndrome: achieving early diagnosis and treatment Am J Kidney Dis Kashtan CE 272279772021 https://doi.org/10.1053/j.ajkd.2020.03.0263271201610.1053/j.ajkd.2020.03.026 · doi ↗ · pubmed ↗
- 4KDIGO 2024 Clinical Practice Guideline for the evaluation and management of chronic kidney disease Kidney Int 03141052024 https://doi.org/10.1016/j.kint.2023.10.01810.1016/j.kint.2023.10.01838490803 · doi ↗ · pubmed ↗
- 5SGLT 2 inhibitors in CKD: are they really effective in all patients?Nephrol Dial Transplant Romagnani P 2025 https://doi.org/10.1093/ndt/gfaf 05110.1093/ndt/gfaf 051PMC 1247747040053507 · doi ↗ · pubmed ↗
- 6Sodium-glucose cotransporter-2 inhibitors in patients with hereditary podocytopathies, alport syndrome, and FSGS: a case series to better plan a large-scale study Cells Boeckhaus J Gross O 10202110.3390/cells 10071815 PMC 830321934359984 · doi ↗ · pubmed ↗
- 7Protocol and rationale for a randomized controlled SGLT 2 inhibitor trial in paediatric and young adult populations with chronic kidney disease: DOUBLE PRO-TECT Alport Nephrol Dial Transplant Gross O Boeckhaus J Weber LT 679687402025 https://doi.org/10.1093/ndt/gfae 1803912265010.1093/ndt/gfae 180PMC 11960741 · doi ↗ · pubmed ↗