Development of a multiplex loop-mediated isothermal amplification (LAMP) method for differential detection of Mycobacterium bovis and Mycobacterium tuberculosis by dipstick DNA chromatography
Mwangala L. Akapelwa, Thoko F. Kapalamula, Lavel C. Moonga, Precious Bwalya, Eddie S. Solo, Joseph Y. Chizimu, Jeewan Thapa, Kyoko Hayashida, Bernard M. Hang'ombe, Musso Munyeme, Aki Tamaru, Takayuki Wada, Shiomi Yoshida, Takuya Kodera, Mistuo Kawase, Stephen V. Gordon

TL;DR
A new low-cost test can quickly tell the difference between two types of tuberculosis bacteria, which is important for proper treatment and tracking spread.
Contribution
A novel multiplex LAMP method with dipstick chromatography for rapid differential detection of M. bovis and M. tuberculosis.
Findings
The multiplex LAMP assay achieved 500 fg and 1 pg DNA sensitivity for M. bovis and M. tuberculosis detection, respectively.
The assay correctly distinguished between M. bovis and M. tuberculosis in 60 minutes with dipstick visualization in 10 minutes.
The method showed high specificity with no cross-reactivity to non-tuberculous mycobacteria or other respiratory pathogens.
Abstract
Although human tuberculosis (TB) caused by Mycobacterium bovis is clinically, pathologically, and radiologically indistinguishable from Mycobacterium tuberculosis-caused TB, M. bovis is innately resistant to pyrazinamide, a key first-line drug effective against M. tuberculosis. The rapid differentiation of these two biovars is therefore of high clinical and epidemiologic importance. Most current molecular tools in resource-limited settings identify mycobacteria only to the M. tuberculosis species (MTB) level. In this study, we report a multiplex loop-mediated isothermal amplification (LAMP) method coupled with dipstick chromatography for the rapid and easy differential detection of M. bovis and M. tuberculosis. The assay was optimized and validated using 143 isolates comprising six MTB reference strains, 50 M. bovis isolates, 58 M. tuberculosis isolates, 24 non-tuberculous mycobacterial…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
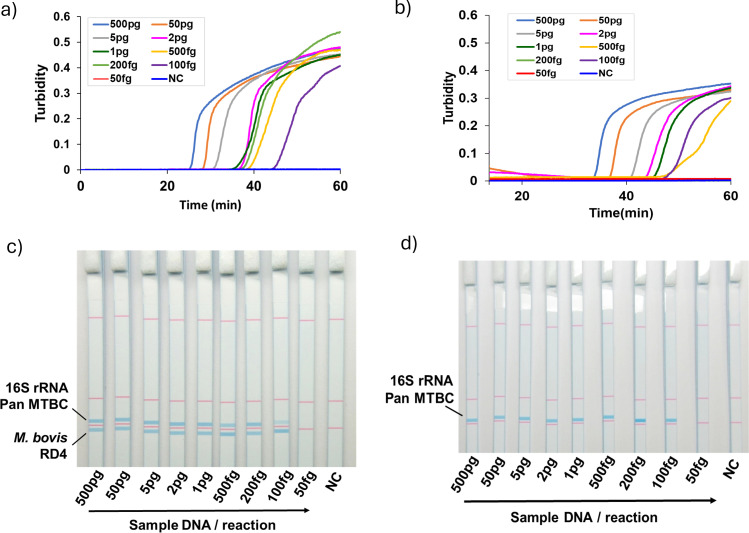 Fig 1
Fig 1| Category | Bacterial species/biovar | Sample ID |
|---|---|---|
| MTB reference strains | Tokyo 172 | |
|
| H37Rv | |
|
| KK 13-02 | |
|
| ATCC 19422 | |
|
| NepR1 | |
|
| EPDC01 | |
| MTB clinical isolates |
| 50 isolates |
|
| 58 isolates | |
| NTM reference strains |
| JATA 51-01 |
|
| KK 44-01 | |
|
| JATA 33-01 | |
|
| JATA 9H-01 | |
|
| JATA 52-01 | |
|
| KK 21-01 | |
|
| JATA 9N-01 | |
|
| KK 24-01 | |
|
| JATA 47-01 | |
|
| JATA 54-01 | |
|
| JATA 43-02 | |
|
| JATA 22-01 | |
|
| JATA 9L-01 | |
|
| JATA 42-01 | |
|
| JATA 23-01 | |
|
| JATA 31-01 | |
|
| JATA 45-01 | |
|
| JATA 50-01 | |
|
| JATA 62-01 | |
|
| JATA 63-01 | |
|
| JATA 61-01 | |
|
| JATA 9P-01 | |
|
| JATA 61-02 | |
|
| JATA 64-01 | |
| Non-mycobacteria |
| NBRC 12689 |
|
| NBRC 100910 | |
|
| NBRC 14401 | |
|
| NBRC 102642 | |
|
| NBRC 3318 |
| Target pathogen | Target gene name | Primer name | Primer sequence name | Primer sequence |
|---|---|---|---|---|
|
| RD4 | RD4-F3 | 5′′- | |
| RD4-B3 | 5′- | |||
| RD4-FIP | 5′- | |||
| RD4-BIP | 5′- | |||
| RD4-LF-Bio | 5′-Bio- | |||
| RD4-LB-TagF1 | 5′-TagF1- | |||
| MTB | 16S rRNA | MTBC | 16 S-F3 | 5′- |
| 16S-B3 | 5′- | |||
| 16S-FIP | 5′- | |||
| 16S-BIP-Bio | 5′-Bio- | |||
| 16S-LF-TagF2 | 5′-TagF2- | |||
| 16S-LB | 5′- |
| Multiplex LAMP-PAS | Multiplex PCR ( | |||||
|---|---|---|---|---|---|---|
| Positive | Positive | |||||
| MTB (16S) | Negative | MTB | Negative | |||
|
| 51 | 51 | 0 | 0 | 51 | 0 |
|
| 59 | 0 | 0 | 59 | 0 | 0 |
| NTM | 0 | 0 | 21 | na | na | na |
|
| 1 | 0 | 0 | |||
|
| 1 | 0 | 0 | |||
|
| 1 | 0 | 0 | |||
| Non-mycobacteria | 0 | 0 | 5 | na | na | na |
- —Ministry of Education, Culture, Sports, Science and Technologyhttp://dx.doi.org/10.13039/501100001700
- —Japan Agency for Medical Research and Developmenthttp://dx.doi.org/10.13039/100009619
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsBiosensors and Analytical Detection · Mycobacterium research and diagnosis · Bacteriophages and microbial interactions
INTRODUCTION
Mycobacterium tuberculosis var. bovis (M. bovis) and Mycobacterium tuberculosis var. tuberculosis (M. tuberculosis) are the etiological agents of bovine tuberculosis (bTB) and tuberculosis (TB) in animals and humans, respectively. While M. tuberculosis causes human TB, M. bovis causes TB in a wide range of animal hosts, including humans. This has made bTB one of the most important zoonotic diseases worldwide of public health concern (1). In 2016, WHO published a rough estimate of the global burden of zoonotic TB, with 147,000 new cases and 12,500 deaths annually (2). Although M. tuberculosis is the main cause of human TB, an estimated 2.8% of new human TB cases in African countries are reportedly caused by M. bovis (3).
Tuberculosis can generally be treated by the WHO-recommended regimen comprising isoniazid (INH), rifampicin (RIF), ethambutol (EMB), and pyrazinamide (PZA). Pyrazinamide is also a key component of drugs recommended for the management of multidrug-resistant (MDR) TB, owing to its ability to act in the acidic environment in which M. tuberculosis persists (4, 5). However, human TB caused by M. bovis cannot be treated with PZA because M. bovis strains are naturally resistant to PZA (6). PZA is known to be the most hepatotoxic of the first-line TB drugs, and its toxicity can be quite severe (4). Therefore, to avoid treatment failures and unnecessary exposure to the potentially toxic drug, there is a need to accurately distinguish M. bovis from M. tuberculosis, especially in areas where bovine and human TB are endemic and coexist. Furthermore, the distinction of M. bovis from M. tuberculosis can assist in monitoring the zoonotic spread of M. bovis to humans, which is important in the formulation of effective control measures (7).
Despite the clinical implications of M. tuberculosis biovar differentiation, it is not routinely performed, partly due to the close genetic relatedness of M. bovis and M. tuberculosis, which makes definitive detection to the biovar level both difficult and time-consuming. Conventional culture and biochemical tests have previously been employed in some studies to differentiate M. bovis from M. tuberculosis. Unfortunately, these methods have the disadvantage of being laborious, slow, and unreliable due to the emergence of strains with variable and intermediate biochemical patterns (7). Nucleic acid amplification tests (NAATs) are a reliable approach for M. bovis from M. tuberculosis differentiation. However, these methods have certain disadvantages, such as high costs of equipment and maintenance, among others, which makes them unfavorable for routine use in resource-limited settings (8, 9).
On the other hand, loop-mediated isothermal amplification (LAMP) is a widely accepted, rapid, low-cost, isothermal DNA amplification technique that utilizes Bst DNA polymerase with strand displacement activity (10). Several highly sensitive LAMP methods have been developed for the detection of M. tuberculosis (11–13), including a method we previously developed for M. bovis detection (14). However, these LAMP systems are limited to detecting a single target due to the use of turbidity and/or fluorescence, which does not allow for differentiation of multiple amplicons. Nevertheless, multiplex LAMP techniques have been established that achieve the simultaneous detection of multiple DNA targets in a single reaction (15). The resultant multiple LAMP products are distinguished by the incorporation of various visualization methods, ranging from fluorophore quencher-based probes to DNA lateral flow dipstick techniques (16–19). Given that the use of fluorescence-based systems requires sophisticated equipment and technical expertise, DNA lateral flow dipstick techniques are a more cost-effective approach (19–21).
Single-stranded tag hybridization chromatographic printed-array strip (STH C-PAS) is a method developed by Tohoku Bio-Array Company Limited (TBA Co., Ltd., Miyagi, Japan), which allows the discrimination of multiple amplified products simultaneously by the principle of DNA-DNA hybridization and DNA lateral flow dipstick (20, 22). In STH C-PAS, target DNA is amplified with a biotin-labeled primer and a unique single-stranded tagged primer. The resultant biotin-labeled amplicons are mixed with streptavidin-coated blue latex beads that form a hybrid due to the strong biotin-streptavidin affinity. When the tip of the dipstick strip (C-PAS) is inserted into the mixture, the amplicon-blue latex hybrids move up through the strip by capillary action and are captured by hybridization to the tag-complementary sequence printed as test lines on the C-PAS (https://www.t-bioarray.com/en_contents/technology.html). The accumulation of the colored amplicons (amplicon-blue latex hybrids) induces a visible blue test line signaling the presence of the target DNA in the sample (23–25). Multiplex DNA signals in a single tube can be easily differentiated and visualized by the C-PAS method within 10 min. Owing to its simplicity, C-PAS has been used as a visualization technology for differential and simultaneous detection of pathogens (21, 23–28) as well as in the food industry (19, 20).
In this report, we describe a low-cost multiplex LAMP-PAS assay for the easy and rapid simultaneous differential detection of M. bovis and M. tuberculosis and applicable for the surveillance of tuberculosis in livestock and humans. The differentiation of M. bovis and M. tuberculosis is essential for the proper management of human TB caused by M. bovis, while routine surveillance of human and livestock TB can aid in monitoring the transmission dynamics of M. bovis from cattle to humans.
MATERIALS AND METHODS
Study samples
A total of 143 mycobacterial and non-mycobacterial strains were used in this study comprising six M. tuberculosis species (MTB) reference strains, non-tuberculous mycobacteria (NTM) reference strains (n = 24), non-mycobacterial respiratory reference strains (n = 5), M. tuberculosis clinical isolates (n = 58), and M. bovis tissue isolates (n = 50). M. bovis BCG Tokyo 172 was provided by the Japan BCG Laboratory (Tokyo, Japan). M. orygis NepR1 and M. caprae EPDC01 were isolated from wild and captive animals and characterized previously (29, 30). Other MTB reference strain samples were provided as extracted and purified DNA by Osaka Institute of Public Health (Osaka, Japan). NTM reference strains were obtained from the Japan Anti-Tuberculosis Association (JATA) Research Institute of Tuberculosis (Tokyo, Japan). Five non-mycobacterial strains were purchased from Biological Resource Center, National Institute of Technology and Evaluation (NBRC) (Chiba, Japan).
Thirty M. bovis samples were collected from cattle suspected of TB during routine postmortem at Lilongwe cold storage abattoir in Lilongwe Malawi in 2019, and the isolates grown in Mycobacterium Growth Indicator Tubes (MGIT) (Becton, Dickinson and Company, NJ, USA) were obtained at National Tuberculosis Reference Laboratory, Lilongwe, Malawi (31). Twenty M. bovis isolates grown on Ogawa medium (Kyokuto Pharmaceutical Industrial Co., Ltd., Tokyo, Japan) were collected from cattle and wild lechwe antelope from 2004 to 2008 in Zambia (32). Twenty-three M. tuberculosis clinical samples grown in MGIT were obtained at the University Teaching Hospital in Lusaka, Zambia during 2014 to 2016 (33). The other 35 M. tuberculosis isolates were collected in Osaka, Japan, during 2000 to 2009 and grown on Ogawa medium at Osaka Institute of Public Health (34). All the used clinical or tissue isolates were confirmed as M. tuberculosis or M. bovis by spoligotyping and multiplex PCR reported by Bakshi et al. (9). Details of the isolates used in this study are outlined in Table 1 and Table S1.
Sample storage and DNA extraction
Reference strains were suspended in 20% glycerol and stored at −80°C at Hokkaido University, Japan. Clinical and tissue isolates were stored in the collaborator’s laboratories, and only extracted DNA samples were transferred to Japan. Genome DNA from the reference strains and Osaka isolates was extracted and purified by the bead-beating method, followed by chloroform purification and ethanol precipitation as previously described (35). For clinical and tissue isolates cultured in liquid MGIT medium, 500 µL of the medium was taken in a cryotube, and crude DNA was extracted by repeated boiling at 95°C for 15 min and freezing at −30°C. For isolates on solid medium, a spoonful colony was suspended in 500 µL of TE buffer in a cryotube, and crude DNA was extracted by repeated boiling and freezing as described above. The final bacterial suspension was centrifuged, and the supernatant was used as the sample. All the extracted DNA samples were kept at −30°C until use.
Multiplex LAMP primers
Two sets of specific primers targeting M. bovis RD4 (14) and the 16S rRNA gene of MTB (12) were synthesized based on our previously reported LAMP protocols. The M. bovis RD4 primers were specific for the RD4 deletion of M. bovis, while 16S rRNA primers targeted all MTB biovars, which include M. bovis. Hence, M. bovis could be positively identified with both primer sets, while other MTB biovars including M. tuberculosis could be identified only by the second primer set. The multiplex LAMP reaction was comprised of two sets of six LAMP primers per target (a total of 12 primers). Two primers from each set underwent some modifications at the 5′ terminal ends. Specifically, the backward loop primer (LB) of the M. bovis RD4 LAMP primer set and the forward loop primer (LF) of the MTB 16S rRNA primer set were tagged with a carbon spacer and a unique tag sequence complementary to the specific oligos printed on the C-PAS (TBA Co., Ltd.) (22). Furthermore, the forward loop primer (LF) and backward inner primer (BIP) targeting M. bovis RD4 and MTB 16S rRNA, respectively, were labeled with biotin at the 5′-terminal (TBA Co., Ltd.). No modifications were made to any of the other primers as outlined in Table 2. All primers without modification were synthesized by Life Technologies Japan Ltd. (Tokyo, Japan).
Multiplex LAMP amplification
The LAMP mixture was the same as previously reported (12, 14)[ however, the LAMP primers were diluted in varying proportions to achieve maximum sensitivity and specificity. The multiplex LAMP reaction was performed in 25 µL reaction volumes as previously described (12). Each reaction contained 2 µL of the DNA template, specific concentrations of primers, 0.8 M betaine (Sigma-Aldrich, St Louis, MO, USA), 20 mM Tris-HCl (pH 8.8) (Wako Pure Chemical Industries, Osaka, Japan), 1.25 mM deoxynucleoside triphosphate mix, 10 mM KCl, 10 mM (NH4)2_SO_4, 0.1% Tween 20 (Sigma-Aldrich), 6 mM MgSO_4_, and 8 U of Bst DNA polymerase (Nippon Gene Co, Ltd, Tokyo, Japan). These reagents were mixed in one tube, and double distilled water (DDW) was added up to a final volume of 25 µL. Thereafter, the mixture was incubated at 66°C in a Loopamp real-time turbidimeter (LA-200; Teramecs Co, Kyoto, Japan) for up to 60 min. The results were considered positive based on the increase in turbidity curves to greater than a threshold of 0.1 according to the manufacturer’s instructions. Extracted DNA from M. bovis BCG Tokyo 172 and M. tuberculosis H37Rv was used as positive control and DDW as a negative control. The reaction mixture was prepared in a clean room before being transferred to the amplification room, where the DNA templates were added.
Visualization of multiplex amplicons by dipstick DNA chromatography (C-PAS)
After the multiplex LAMP reaction, the C-PAS F4 membrane strip (TBA Co., Ltd.) was inserted into a 33 µL reaction mix containing 30 µL of developing solution (300 mM NaCl) (TBA Co., Ltd.), 2 µL of streptavidin-coated blue latex suspension (TBA Co., Ltd.), and 1 µL of LAMP product. The strip was incubated for 10 min at room temperature, after which the results were interpreted. A positive test was indicated by the appearance of a blue line on the C-PAS strip test position signaling the presence of the amplified target DNA sequence tagged with the complementary oligonucleotide and biotin. The best combination of modified primers and the ratio of each primer were determined from the coloration of the bands of interest. The LAMP amplification and the visualization by dipstick chromatography were performed in the post-amplification room, which is separate from the pre-amplification clean room.
Specificity analysis
The multiplex LAMP specificity was evaluated against MTB reference strains, NTM strains, non-mycobacterial respiratory strains, M. tuberculosis clinical isolates, and M. bovis tissue isolates (Table 1; Table S1) in comparison with the multiplex PCR assay targeting RD4 described by Bakshi et al. (9). The multiplex PCR procedure was slightly modified. Briefly, 4 µL 5× Go Taq buffer (Promega Co., WI, USA); 0.8 µL of 25 mM MgCl_2_; 0.2 µL of 25 mM each dNTP mix; 2 µL of 5 M betaine, 0.5 µL of 10 µM each primer; common forward primer - CBS1 (5′-TTCCGAATCCCTTGTGA-3′); *M. bovis-*specific reverse primer—CBS2 (5′-GGAGAGCGCCGTTGTA-3′), and *M. tuberculosis-*specific reverse primer—CBS3 (5′-AGTCGCCGTGGCTTCTCTTTTA-3′); 0.1 µL of 5 U/µL GoTaq DNA polymerase (Promega Co.); 1 µL of template DNA; and finally DDW to make up to a final volume of 20 µL. The cycling parameters consisted of an initial denaturation at 94°C for 5 min, followed by 30 cycles of denaturation at 94°C (1 min), annealing at 52°C (1.5 min), and extension at 72°C (1 min), with a final elongation step at 72°C for 5 min. The amplification products were analyzed by gel electrophoresis at 100 V for 20 min. The predicted PCR products were 168 bp (M. bovis) and 268 bp (MTB other than M. bovis).
Evaluation of the limit of detection (LoD)
The LoD of our multiplex LAMP system was evaluated using serially diluted M. bovis BCG Tokyo 172 and M. tuberculosis H37Rv genomic DNA, ranging from 250 pg/µL to 25 fg/μL (500 pg, 50 pg, 5 pg, 2 pg, 1 pg, 500 fg, 200 fg, 100 fg, and 50 fg/reaction). The DNA concentration was measured using Qubit 3 Fluorometer (Thermo Fisher Scientific, MA, USA) according to the manufacturer’s instructions. Reactions were performed multiple times as shown in Table S2.
RESULTS
Rapid differential detection of M. bovis and M. tuberculosis by multiplex LAMP
Multiplex LAMP reaction was optimized for M. bovis and M. tuberculosis detection by utilizing a Loopamp real-time turbidimeter coupled with C-PAS. The following concentrations were selected for each primer after examining various concentrations: 1.6 µM FIP/BIP, 0.8 µM LF/LB, 0.2 µM F3/B3 of RD4 LAMP primer set, and for MTB LAMP primer set, 0.8 µM FIP/BIP, 0.4 µM LF/LB, and 0.1 µM F3/B3. The optimal incubation temperature and time were 66°C and 60 min, respectively. The multiplex LAMP reaction results were ready within 60 min, while visualization of results by dipstick chromatography took 10 min.
Specificity of the multiplex LAMP
The specificity of the multiplex LAMP assay was evaluated with strains shown in Table 1 and Table S1. In all cases, reaction mixtures with the template DNAs from M. bovis isolates showed two positive bands on the C-PAS indicating the presence of both targets (*M. bovis-*specific RD4 and MTB 16S rRNA) in the tested samples. On the other hand, all reactions with the DNAs from M. tuberculosis isolates and MTB strains other than M. bovis consistently showed only one positive band on the C-PAS, which signaled the detection of the target MTB 16S rRNA in the test samples (Fig. 1 and Table 3; Table S1). All the results were concordant with those of the multiplex PCR (9), namely, M. bovis showed a band of 168 bp, and MTB other than M. bovis was 268 bp. While the assay successfully detected all M. bovis, M. tuberculosis, and MTB strains, no positive test bands, i.e., no amplification occurred, were observed with any of the non-mycobacterial strains tested. Of the 24 NTM tested, M. marinum, M. ulcerans, and M. asiaticum showed a positive band for the 16S rRNA gene (Table 3). A multiple alignment comparison of the target region of the 16S rRNA gene of these three species indicated high sequence identity to that of MTB (Supplementary Figure). These findings indicate that the two sets of primers utilized in this study are highly specific for the detection of M. bovis and MTB, though there were a few exceptions.
Multiplex LAMP amplification and discrimination by dipstick DNA chromatography (C-PAS). (a, b) Multiplex LAMP results for M. bovis BCG (a) and M. tuberculosis H37Rv (b) detection by rising curves from the turbidimeter. (c, d) Results of M. bovis BCG (c) and M. tuberculosis H37Rv (d) detection by multiplex LAMP coupled with C-PAS. Upper band is for the detection of MTB 16S rRNA, and the lower band is for the detection of M. bovis specific RD4. M. bovis shows two bands, while other MTB shows only one upper band. NC: negative control
LoD of the multiplex LAMP
To estimate the LoD of the multiplex LAMP assay, 10-fold serial dilutions and additional concentrations of each of M. bovis BCG Tokyo 172 and M. tuberculosis H37Rv genomic DNA were used. The multiplex LAMP was able to detect as low as 500 fg of both M. bovis and M. tuberculosis genomic DNA per reaction within 60 min and correctly differentiated them when the concentration of DNA was 1 pg/reaction or higher (Fig. 1; Table S2). The amplicons could be easily differentiated by C-PAS in 10 min (Fig. 1C and D). Since concentrations lower than 500 fg/reaction were sometimes not detected as positive and one of the two bands missed in BCG samples, we decided to stop the incubation at 60 min and 1 pg/reaction was set as the differential detection limit.
DISCUSSION
Mycobacterium tuberculosis is known to be the main cause of human tuberculosis; however, 10% to 30% of human TB cases in some African countries are reportedly caused by M. bovis (3). Despite this, M. bovis differentiation is still not routinely performed due to diagnostic limitations, risking inappropriate case management and poor treatment outcomes. Therefore, assays that can accurately distinguish between these mycobacteria will improve treatment regimens and the establishment of adequate public health control measures. Here, we report a simple multiplex LAMP coupled with dipstick chromatography that allows the rapid identification of M. bovis and other MTB. This multiplex LAMP method is advantageous as it can easily achieve simultaneous amplification of multiple target sequences in a single reaction, which saves time and effort.
While many LAMP methods have been reported to specifically detect M. tuberculosis or MTB (11, 12, 36), there are few methods to specifically detect M. bovis, including PCR methods (9, 14). This is because there are virtually no major variations, such as genes or genomic regions unique to M. bovis, that would allow for differentiation from other MTB strains (37). MTB strains do not undergo horizontal gene transfer, with the phylogeny marked by genomic deletions, making it difficult to find genomic regions that are retained only by a particular biovar. Several biovar-specific point mutations have been reported across the MTB; however, aside from PCR methods, current technology has not been able to reproducibly identify these point mutations using isothermal amplification methods (25).
Based on the above, presuming the use of field-available isothermal amplification methods, the most reliable M. bovis identification region to date is RD4, which was reported by Gordon et al. in 1999 (37, 38). This region is absent in all M. bovis strains known to date, and its length of 12.7 kbp is sufficient to prevent nonspecific amplification in LAMP methods with primer sets designed across the deletion site (14, 37). On the other hand, the 16S ribosomal RNA gene is the reference gene for bacterial species identification, and the variable region we have employed is an ideal target for MTB identification as it is distinguishable from other bacterial species, yet perfectly matched within MTB strains (12). In the present study, three NTM species, M. marinum, M. ulcerans, and M. asiaticum, showed positive results in the detection of MTB 16S rRNA gene due to the high similarity of the sequence to that of MTB in the region (Fig. S1). However, since the pathogenesis and epidemiology of M. marinum and M. ulcerans are different from those of MTB (39, 40) and M. asiaticum is rarely isolated from clinical specimens (41), we do not believe this will be a cause for confusion in the species determination of MTB. Although the LoD of the multiplex LAMP method was slightly higher than that of each LAMP method alone (12, 14), 1 pg of DNA is equivalent to 200 MTB bacilli, which is sensitive enough for detection while discriminating between the biovars.
The LAMP is an excellent isothermal amplification method that can be achieved with a single enzyme, Bst polymerase (10). However, using amplification alone to call positive or negative results has the drawback that nonspecific amplification cannot be distinguished. Although there have been several ways to identify two or more amplifications occurring simultaneously, such as using a fluorescence-quenching system (16, 18), the simplest and most practical method would be to use a lateral flow strip and capture the amplifications in separate bands. In this method, only amplified products sandwiched between two specific sequences are detected, thus avoiding detection of nonspecific amplification. Several researchers have published methods using immunochromatography; however, their stability and inexpensive mass production remain questionable due to the use of antibodies as capturing agents (42, 43). Since STH C-PAS uses single-stranded DNA as the capture, the target can be captured by simply adding a complementary sequenced DNA tag to it. Like DNA arrays, multiple captures can be placed on the same strip, allowing for future applications such as additional targets (19–21, 23–28).
A drawback of the current study is that it did not use specimens collected directly from patients’ sputum or bovine TB lesions prior to culture. Since such specimens are not available in bovine TB-free countries like Japan, we plan to bring this system to endemic countries and conduct additional testing using specimens obtained from hospitals and slaughterhouses. In addition, the cold chain problem needs to be solved in order to implement the project in rural areas where infrastructure development has not caught up and cooling systems are not available. Since both LAMP systems used in this study have been successfully made into dried LAMP systems individually, we plan to dry both systems in a mixed state to confirm their stability and reproducibility (44–46). Once this multiplex-LAMP system can be successfully dried, it can be implemented using only a water bath.
Presently, the true burden of TB due to M. bovis remains largely unknown. This multiplex LAMP-PAS can be applied as an epidemiological tool to investigate the prevalence of TB caused by M. bovis. This assay can also be used for the general detection of MTB members, including M. tuberculosis, which are susceptible to PZA. Most importantly, the rapid differentiation of M. bovis and M. tuberculosis will allow the administration of suitable treatment regimens and the formulation of appropriate public health control measures.
In conclusion, we have developed a simple and rapid multiplex LAMP-PAS system for the simultaneous and differential detection of M. bovis and M. tuberculosis. The accuracy, ease, and low-cost qualities of this assay make it suitable for use at point-of-care settings, especially in resource-poor countries, which have high tuberculosis burdens in both humans and animals. The rapid and accurate differential diagnosis of M. bovis and M. tuberculosis by this multiplex LAMP-PAS technique will facilitate early diagnosis, which is critical for the management and control of tuberculosis in both humans and animals.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Taye H, Alemu K, Mihret A, Wood JLN, Shkedy Z, Berg S, Aseffa A. 2021. Global prevalence of Mycobacterium bovis infections among human tuberculosis cases: systematic review and meta-analysis. Zoonoses Public Health 68:704–718. doi:10.1111/zph.1286834169644 PMC 8487997 · doi ↗ · pubmed ↗
- 2World Health Organization. 2017. Roadmap for zoonotic tuberculosis. Available from: https://iris.who.int/bitstream/handle/10665/259229/9789241513043-eng.pdf?sequence=1
- 3Müller B, Dürr S, Alonso S, Hattendorf J, Laisse CJM, Parsons SDC, van Helden PD, Zinsstag J. 2013. Zoonotic Mycobacterium bovis-induced tuberculosis in humans. Emerg Infect Dis 19:899–908. doi:10.3201/eid 1906.12054323735540 PMC 4816377 · doi ↗ · pubmed ↗
- 4World Health Organization. 2014. Companion handbook to the WHO guidelines for the programmatic management of drug-resistant tuberculosis. Available from: https://apps.who.int/iris/bitstream/handle/10665/130918/?sequence=125320836 · pubmed ↗
- 5Shi W, Zhang X, Jiang X, Yuan H, Lee JS, Barry CE 3rd, Wang H, Zhang W, Zhang Y. 2011. Pyrazinamide inhibits trans-translation in Mycobacterium tuberculosis. Science 333:1630–1632. doi:10.1126/science.120881321835980 PMC 3502614 · doi ↗ · pubmed ↗
- 6Konno K, Feldmann FM, Mc Dermott W. 1967. Pyrazinamide susceptibility and amidase activity of tubercle bacilli. Am Rev Respir Dis 95:461–469. doi:10.1164/arrd.1967.95.3.4614225184 · doi ↗ · pubmed ↗
- 7Allix-Béguec C, Fauville-Dufaux M, Stoffels K, Ommeslag D, Walravens K, Saegerman C, Supply P. 2010. Importance of identifying Mycobacterium bovis as a causative agent of human tuberculosis. Eur Respir J 35:692–694. doi:10.1183/09031936.0013730920190335 · doi ↗ · pubmed ↗
- 8Case RJ, Boucher Y, Dahllöf I, Holmström C, Doolittle WF, Kjelleberg S. 2007. Use of 16S r RNA and rpo B genes as molecular markers for microbial ecology studies. Appl Environ Microbiol 73:278–288. doi:10.1128/AEM.01177-0617071787 PMC 1797146 · doi ↗ · pubmed ↗