Genome assembly of the winter ant, Prenolepis imparis
Elizabeth I Cash, Philip S Ward, Merly Escalona, Ruta Sahasrabudhe, Courtney Miller, Erin Toffelmier, Colin Fairbairn, William Seligmann, H Bradley Shaffer, Neil D Tsutsui

TL;DR
Researchers assembled the genome of the winter ant, Prenolepis imparis, to better understand its cold adaptation and genetic diversity.
Contribution
The study provides a high-quality genome assembly of Prenolepis imparis, enabling future research on thermal adaptation and resilience to climate change.
Findings
The genome assembly consists of 787 scaffolds spanning 327.3 Mb with high contig and scaffold N50 values.
The assembly has a BUSCO completeness of 96.5%, indicating high quality.
The genome will support studies on thermal physiology and adaptation in a warming climate.
Abstract
The winter ant, Prenolepis imparis, is one of the most common, widespread, and conspicuous ant species in North America. P. imparis is well adapted to cold climates, and consequently, is often noted as the only active ant species during colder months. This specialized life history makes P. imparis a useful model organism for exploring thermal physiology and understanding the potential impacts of a warming climate on insects. Phylogeographic studies have revealed deeply divergent lineages across North America, as well as a single collection of an apparent social parasite in California. In light of its distinctive cold adaptation and recently discovered geographic diversity, a better understanding of the underlying genetic patterns of the winter ant is valuable to future conservation efforts for this species. Here, we present a high-quality genome assembly of P. imparis from Santa Clara…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
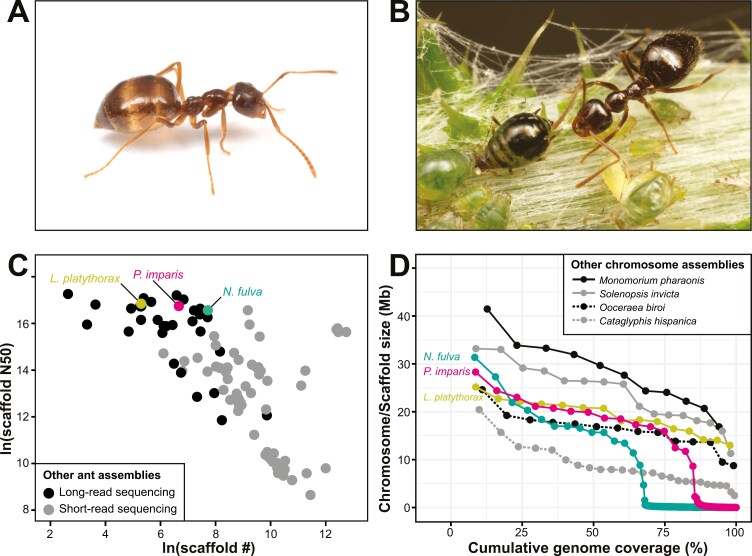 Fig. 1
Fig. 1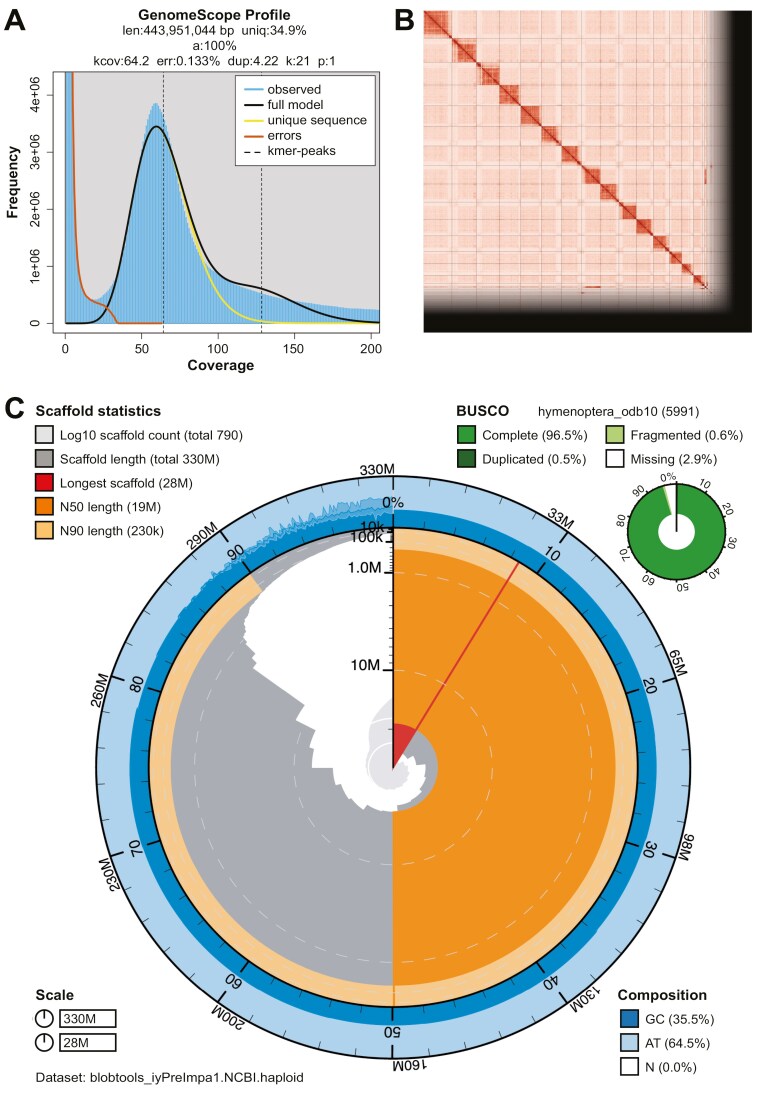 Fig. 2
Fig. 2| Assembly | Software and any non-default options | Version |
|---|---|---|
| Filtering PacBio HiFi adapters | HiFiAdapterFilt | Commit 64d1c7b |
| K-mer counting | Meryl ( | 1 |
| Estimation of genome size | GenomeScope | 2 |
| De novo assembly (contiging) | HiFiasm (--n-hap 1 -l0) | 0.16.1-r375 |
|
| ||
| Omni-C data alignment | Arima Genomics Mapping Pipeline | Commit 2e74ea4 |
| Omni-C Scaffolding | SALSA (-DNASE, -i 20, -p yes) | 2 |
| Gap closing | YAGCloser (-mins 2 -f 20 -mcc 2 -prt 0.25 -eft 0.2 -pld 0.2) | Commit 0e34c3b |
|
| ||
| Short-read alignment | BWA-MEM (-5SP) | 0.7.17-r1188 |
| SAM/BAM processing | samtools | 1.11 |
| SAM/BAM filtering | pairtools | 0.3.0 |
| Pairs indexing | pairix | 0.3.7 |
| Matrix generation | cooler | 0.8.10 |
| Matrix balancing | hicExplorer (hicCorrectmatrix correct --filterThreshold -2 4) | 3.6 |
| Contact map visualization | HiGlass | 2.1.11 |
| PretextMap | 0.1.4 | |
| PretextView | 0.1.5 | |
| PretextSnapshot | 0.0.3 | |
| Manual curation tools | Rapid curation pipeline (Wellcome Trust Sanger Institute, Genome Reference Informatics Team) | Commit 4ddca450 |
|
| ||
| Basic assembly metrics | QUAST (--est-ref-size) | 5.0.2 |
| Assembly completeness | BUSCO (-m geno, -l hymenoptera) | 5.0.0 |
| Merqury | 2020-01-29 | |
|
| ||
| Local alignment tool | BLAST + (-db nt, -outfmt “6 qseqid staxids bitscore std,” -max_target_seqs 1, -max_hsps 1, -evalue 1e-25) | 2.10 |
| General contamination screening | BlobToolKit (PacBIo HiFi Coverage, NCBI Taxa ID = 262038, BUSCODB = Hymenoptera) | 2.3.3 |
|
| ||
| Mitochondrial genome assembly | MitoHiFi (-r, -p 80, -o 1 -a animal) Reference genome: | 2.2 |
|
| ||||||
| CCGP NCBI BioProject | PRJNA720569 | |||||
| Genera NCBI BioProject | PRJNA765835 | |||||
| Species NCBI BioProject | PRJNA808363 | |||||
| NCBI BioSample | SAMN35821884, SAMN35821917 | |||||
| Specimen identification | NDT837.1, NDT839.2m4 | |||||
|
| ||||||
| Assembly accession | GCA_030444845.1 | |||||
| Genome sequences | ||||||
|
| ||||||
| PacBio HiFi reads | Run | 1 PACBIO_SMRT (Sequel II) run: 3.2M spots, 29.2G-bases, 14.4Gb | ||||
| Accession | SRX21253660 | |||||
| Omni-C Illumina reads | Run | 2 ILLUMINA (Illumina NovaSeq 6000) runs: 54.9M spots, 16.6G-bases, 5.6Gb | ||||
| Accession | SRX21253661, SRX21253662 | |||||
|
| ||||||
| Assembly identifier (Quality code | iyPreImpa1(5.7.P.Q56.C86) | |||||
| HiFi Read coverage | 65.73X | |||||
| Number of contigs | 1,480 | |||||
| Contig N50 (bp) | 901,923 | |||||
| Contig NG50 | 338,411 | |||||
| Longest contigs | 12,003,945 | |||||
| Number of scaffolds | 787 | |||||
| Scaffold N50 | 18,679,404 | |||||
| Scaffold NG50 | 16,868,572 | |||||
| Largest scaffold | 28,297,219 | |||||
| Size of final assembly (bp) | 327,284,298 | |||||
| Phased block NG50 | 338,411 | |||||
| Gaps per Gbp (#Gaps) | 2117(693) | |||||
| Indel QV (Frame shift) | 43.84675244 | |||||
| Base-pair QV | 56.5 | |||||
| k-mer completeness | 99.07 | |||||
| BUSCO completeness |
|
|
|
|
| |
| 96.50% | 96.10% | 0.50% | 0.60% | 2.80% | ||
| Organelles (complete mitochondrial sequence) | Size (bp) | 18,821 | ||||
| Accession | ||||||
- —NIH Shared Instrumentation
- —Vincent J. Coates Genomics Sequencing Laboratory
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsInsect and Arachnid Ecology and Behavior · Plant and animal studies · Neurobiology and Insect Physiology Research
Introduction
The winter ant, Prenolepis imparis, is a common and widely distributed ant in North America, with a range that extends throughout most of the continental United States and scattered localities in Mexico and from sea level up to 8,000 feet elevation (Wheeler 1930). The species, as currently defined, includes at least 5 highly divergent lineages, plus a possible microgyne social parasite known from a single collection in California (A.L. Wild, personal communication; Tonione et al. 2022). As its name suggests, the winter ant is a cold-adapted species, commonly active during the cooler months of the year or when daytime temperatures are too cold for other ant species. Colonies are often found in cool, shady, and moist microhabitats (Wheeler 1930). Additionally, P. imparis exhibits distinctive biological traits such as storing fat and other nutrients in specialized “replete” workers, which act as living energy reserves for the colony during the annual brood-rearing cycle (Tschinkel 1987, Fig. 1A), and the formation of mutualistic relationships with aphids and scale insects (Fig. 1B). These and other features highlight its ecological adaptability and complex social behaviors (Wheeler 1930).
Winter ant, P. imparis, and genome assembly comparisons. (A) Representative of P. imparis showing a replete worker with nutrient stores in gaster (image credit: Elizabeth I. Cash). (B) Example of a P. imparis worker (top right) tending aphid mutualists (bottom left) (image credit: Elizabeth I. Cash). (C) Scatterplot of genome metrics for 97 ant genomes representing 72 species with scaffold-level assemblies accessed via NCBI Datasets (see Supplementary Table S1 for full details). Three focal species, P. imparis (this study, long-read sequencing), L. platythorax (Feldmeyer et al. 2023, long-read sequencing), and N. fulva (Allen et al. 2023, long-read sequencing), are highlighted to compare scaffold assemblies of these closely related taxa with 94 other ant genome assemblies (shaded according to sequencing method, i.e, long- versus short-read lengths). (D) Lineplot of genome metrics comparing 6 ant species with P. imparis. The size of each chromosome or scaffold (Mb) is displayed on the y-axis and color formatted by species. The cumulative genome coverage (%) is represented on the x-axis by the sum of the preceding chromosome or scaffold size(s) up to a given point. Chromosome-level assemblies for 6 ant species (C. hispanica, L. platythorax, M. pharaonis, N. fulva, O. biroi, and S. invicta) are compared with the winter ant, P. imparis (this study) showing the similarity between assembled scaffold sizes of P. imparis and the chromosome sizes of 2 closely related Lasiini species (L. platythorax and N. fulva; see Supplementary Table S2 for full details).
Cold-adapted organisms, including P. imparis, are especially vulnerable to a warming climate, and are thus valuable models for understanding how individuals, populations, and species might be affected by climate change. Tests of thermal physiology in P. imparis have shown that populations in California vary in their resistance to both heat and cold (Tonione et al. 2020b), and that over 600 genes are differentially expressed in response to heat shock, whereas only 7 genes exhibit significant changes following cold shock (Tonione et al. 2020a). The insights gained from these transcriptomic findings underscore the importance of establishing a reference genome for P. imparis, which will serve as a crucial resource for understanding the genetic basis of thermal tolerance and adaptation in the face of climate change.
Here, we report a high-quality de novo genome assembly for P. imparis collected in Santa Clara County, California. Existing genomic resources for P. imparis include the aforementioned transcriptome sequences and ultraconserved element sequences of specimens from throughout North America (Tonione 2022). Additional genomic resources for closely related species in the tribe Lasiini include Lasius niger, Lasius platythorax, and Nylanderia fulva. We highlight comparisons between P. imparis (this study) and the whole genome sequences of these 3 Lasiini species, as well as the whole genome sequences of other ant species currently available in the NCBI genome database (Fig. 1C and D). As part of the California Conservation Genomics Project (CCGP), this genome assembly will be a foundational resource for broader population genomic studies that will allow us to define taxa, populations, and regions of high conservation concern (Fiedler et al. 2022; Shaffer et al. 2022).
Methods
Biological materials
In February 2021, queen P. imparis ants were collected during annual nuptial flights in Santa Clara County, California and reared in the lab. To approximate colony-founding conditions, queens were kept individually in water-filled glass tubes stoppered with cotton plugs in ambient temperature conditions (24–26 °C) and total darkness. Queens were monitored once a week for the presence of brood, and male offspring were collected as they developed into pupae or newly eclosed adults. Live samples were flash frozen with liquid nitrogen and stored at –80 °C until processed. DNA for HiFi SMRTbell library construction and sequencing was extracted from 1 adult male P. imparis (collection code NDT839.2.m4, NCBI BioSample SAMN35821917) that was produced in captivity by a queen collected from a nuptial flight in Linda Vista Park in Cupertino, Santa Clara County, California (N 37.306842, W –122.060485) on 23 February 2021. DNA for Omni-C library construction and sequencing was extracted from 3 male P. imparis pupae (collection codes NDT837.1.m1, NDT837.1.m2, NDT837.1.m3, listed collectively under NCBI Specimen Name NDT837.1, NCBI BioSample SAMN35821884) that were produced in captivity by a single queen collected in February 2021 from Serra Park in Sunnyvale, Santa Clara County, California (N 37.343696, W –122.042545). Given this, the genome assembly was produced from 4 haploid (male) individuals, originating from 2 P. imparis populations that were 4.4 km apart.
High molecular weight genomic DNA preparation
A flash-frozen adult male ant (NDT839.2.m4) was homogenized in 650 µL of homogenization buffer (10 mM Tris-HCL-pH 8.0 and 25 mM EDTA) using TissueRuptor II (Qiagen, Germany; Cat # 9002755). Lysis buffer (650 µL, 10 mM Tris, 25 mM EDTA, 200 mM NaCl, and 1% SDS) and proteinase K (100 µg/mL) were added to the homogenate and incubated overnight at room temperature. Lysate was treated with RNAse A (20 µg/mL) at 37 °C for 30 min and was cleaned with equal volumes of phenol/chloroform using phase-lock gels (Quantabio, Beverly, MA; Cat # 2302830). The DNA was precipitated by adding 0.4 × volume of 5M ammonium acetate and 3 × volume of ice-cold ethanol. The DNA pellet was washed twice with 70% ethanol and resuspended in an elution buffer (10 mM Tris, pH 8.0). DNA yield was 20 ng total as measured by the Qubit 2.0 Fluorometer (Thermo Fisher Scientific, Waltham, MA). Integrity of the high molecular weight genomic DNA (HMW gDNA) was verified on a Femto pulse system (Agilent Technologies, Santa Clara, CA), where 54% of DNA was observed in fragments larger than 50 kb.
The HiFi SMRTbell library was constructed using the SMRTbell gDNA Sample Amplification Kit (Pacific Biosciences [PacBio], Menlo Park, CA; Cat. #101-980-000) and the SMRTbell Express Template Prep Kit 2.0 (PacBio; Cat. #100-938-900) according to the manufacturer’s instructions. High molecular weight genomic DNA was sheared to approximately 10 kb using the Megaruptor 3 system (Diagenode, Belgium; Cat. #B06010003). Sheared genomic DNA was incubated at 37 °C for 15 min to remove single-strand overhangs, followed by DNA damage repair at 37 °C for 30 min, end repair and A-tailing at 20 °C for 30 min and 65 °C for 30 min, and ligation of amplification adapters at 20 °C for 60 min. To prepare for library amplification by PCR, the library was purified with ProNex beads (Promega, Madison, WI; Cat. # NG2002) for 2 PCR amplification conditions at 15 cycles each then another ProNex bead purification. Purified amplified DNA from both reactions was pooled in equal mass quantities for another round of enzymatic steps that included DNA repair, end repair/A-tailing, overhang adapter ligation, and purification with ProNex Beads. The PippinHT system (Sage Science, Beverly, MA; Cat # HPE7510) was used for SMRTbell library size selection to remove fragments < 6 kb. The 10 kb average HiFi SMRTbell library was sequenced at UC Davis DNA Technologies Core (Davis, CA) using 1 8M SMRT cell, Sequel II sequencing chemistry 2.0, and 30-h movies each on a PacBio Sequel II sequencer.
Omni-C preparation
The Omni-C library was prepared using the Dovetail Omni-C Kit (Dovetail Genomics, Scotts Valley, CA) according to the manufacturer’s protocol with slight modifications. First, specimen tissue (whole individuals, 3 male pupae, specimen numbers NDT837.1.m1, NDT837.1.m2, and NDT837.1.m3) was thoroughly ground with a mortar and pestle while cooled with liquid nitrogen. Subsequently, chromatin was fixed in place in the nucleus. The suspended chromatin solution was then passed through 100 μm and 40 μm cell strainers to remove large debris. Fixed chromatin was digested under various conditions of DNase I until a suitable fragment length distribution of DNA molecules was obtained. Chromatin ends were repaired and ligated to a biotinylated bridge adapter followed by proximity ligation of adapter-containing ends. After proximity ligation, cross-links were reversed and the DNA was purified from proteins. Purified DNA was treated to remove biotin that was not internal to ligated fragments. An NGS library was generated using an NEB Ultra II DNA Library Prep kit (New England Biolabs, Ipswich, MA) with an Illumina compatible y-adaptor. Biotin-containing fragments were then captured using streptavidin beads. The post-capture product was split into 2 replicates prior to PCR enrichment to preserve library complexity with each replicate receiving unique dual indices. The library was sequenced at Vincent J. Coates Genomics Sequencing Lab (Berkeley, CA) on an Illumina NovaSeq 6000 platform (Illumina, CA) to generate approximately 100 million 2 × 150 bp read pairs per GB of genome size.
Genome assembly
Nuclear genome assembly
We assembled the P. imparis genome following the CCGP assembly pipeline for haploid genomes, as outlined in Table 1, which lists the tools and non-default parameters used in the assembly. The pipeline uses PacBio HiFi reads and Omni-C data to produce high-quality and highly contiguous genome assemblies. First, we removed the remnant adapter sequences from the PacBio HiFi dataset using HiFiAdapterFilt (Sim et al. 2022) and generated an initial haploid assembly using HiFiasm (Cheng et al. 2022) with the filtered PacBio HiFi reads, specifying no purging and the ploidy corresponding to the sequenced individual, a haploid male. From the generated output, we kept the file corresponding to the primary assembly file. We then aligned the Omni-C data to the assembly following the Arima Genomics Mapping Pipeline (https://github.com/ArimaGenomics/mapping_pipeline) and then scaffolded it with SALSA (Ghurye et al. 2017, 2019).
The assembly was manually curated by generating and analyzing its corresponding Omni-C contact maps and breaking scaffolds when misassemblies were identified. We aligned the Omni-C data with BWA-MEM (Li 2013) to generate the contact maps and identified ligation junctions to generate Omni-C pairs (Lee et al. 2022) using pairtools (Open2C et al. 2024). We generated multi-resolution Omni-C matrices with cooler (Abdennur and Mirny 2020) and balanced them with hicExplorer (Ramírez et al. 2018). We used HiGlass (Kerpedjiev et al. 2018) and the PretextSuite (https://github.com/wtsi-hpag/PretextView; https://github.com/wtsi-hpag/PretextMap; and https://github.com/wtsi-hpag/PretextSnapshot) to visualize the contact maps where we identified misassemblies and misjoins. Some of the remaining gaps (joins generated during scaffolding and/or curation) were closed using the PacBio HiFi reads and YAGCloser (https://github.com/merlyescalona/yagcloser). Finally, we checked for contamination using the BlobToolKit Framework (Challis et al. 2020).
Genome quality assessment
We generated k-mer counts from the PacBio HiFi reads using meryl (https://github.com/marbl/meryl). The k-mer counts were then used in GenomeScope2.0 (Ranallo-Benavidez et al. 2020) to estimate genome features including genome size, heterozygosity, and repeat content. To obtain general contiguity metrics, we ran QUAST (Gurevich et al. 2013). To evaluate genome quality and functional completeness, we used BUSCO (Manni et al. 2021) with both Hymenoptera ortholog databases (hymenoptera_odb10) that contain 5,991 genes. Assessment of base level accuracy (QV) and k-mer completeness was performed using the previously generated meryl database and merqury (Rhie et al. 2020). We further estimated genome assembly accuracy via BUSCO gene set frameshift analysis using the pipeline described in Korlach et al. (2017). Given that the specimen used for the assembly is haploid, measurements of the size of the phased blocks are based on the size of the final contigs. We follow the quality metric nomenclature established by Rhie et al. (2021), with the genome quality code x.y.P.Q.C, where, x = log10[contig NG50]; y = log10[scaffold NG50]; P = log10 [phased block NG50]; Q = Phred base accuracy QV (quality value); C = % genome represented by the first “n” scaffolds, following an estimated karyotype for Prenolepis of n = 16 inferred from Omni-C contact map results (Fig. 2B) and previous reports of the number of chromosomes for the genus (Goni et al. 1982; Imai et al. 1983).
Visual overview of P. imparis genome assembly metrics. (A) K-mer spectrum output generated from PacBio HiFi data without adapters using GenomeScope 2.0. The observed unimodal pattern corresponds to a haploid genome. (B) Omni-C contact map for the scaffold-level genome assembly generated with PretextSnapshot. The Omni-C contact map translates the proximity of genomic regions in 3D space to contiguous linear organization. Each cell in the contact map corresponds to sequencing data supporting the linkage (or join) between 2 such regions. Scaffolds are separated by black lines, with higher density corresponding to higher levels of fragmentation. (C) BlobToolKit Snail plot showing a graphical representation of the quality metrics presented in Table 2 for the P. imparis assembly (iyPreImpa1) and BUSCO assessment results based on the Hymenoptera set of orthologous genes (n = 5,991). The plot circle represents the full size of the assembly. From the inside to the outside, the central plot covers length-related metrics. The red line represents the size of the longest scaffold; all other scaffolds are arranged in size order moving clockwise around the plot and drawn in gray starting from the outside of the central plot. Dark and light orange arcs show the scaffold N50 and scaffold N90 values, respectively. The central light gray spiral shows the cumulative scaffold count with a white line at each order of magnitude. White regions in this area reflect the proportion of Ns in the assembly. The dark versus light blue area around it shows mean, maximum, and minimum GC vs. AT content at 0.1% intervals.
Mitochondrial genome assembly
We assembled the mitochondrial genome of P. imparis from the PacBio HiFi reads using the reference-guided pipeline MitoHiFi (Allio et al. 2020; Uliano-Silva et al. 2023). The mitochondrial sequence of Lasius spathepus (NCBI:NC_053901.1) was used as the starting sequence. After completion of the nuclear genome, we searched for matches of the resulting mitochondrial assembly sequence in the nuclear genome assembly using BLAST+ (Camacho et al. 2009) and filtered out contigs and scaffolds from the nuclear genome with a sequence identity >99% and size smaller than the mitochondrial assembly sequence. No other manual curation was performed on the mitochondrial genome.
Genome assembly comparisons
We compared basic scaffold-level assembly metrics for 97 ant genomes representing 72 ant species currently available in the NCBI genome database (Supplementary Table S1). Scaffold number versus scaffold N50 (ln transformed) were plotted using ggplot2 in R (Wickham 2016) to visualize differences in contiguity between ant genome assemblies (Fig. 1C). Additionally, scaffold and chromosome sizes (Mb) were plotted relative to genome coverage (%) for 8 ant species including chromosome-level assemblies of 4 non-Lasiini species (Cataglyphis hispanica, Monomorium pharaonis, Ooceraea biroi, and Solenopsis invicta) and 2 Lasiini species (N. fulva and L. platythorax), as well as scaffold-level assemblies of 2 Lasiini species (L. niger and P. imparis [this study]), to compare mapping results among genome assemblies (Fig. 1D, see Supplementary Table S1 for accession numbers and references, and Supplementary Table S2 for data).
Results
Sequencing data
The Omni-C and PacBio HiFi sequencing libraries generated 54.87 million read pairs and 3.17 million reads respectively. The latter yielded 65.73-fold coverage and had an N50 read length 9167 bp; minimum read length 123 bp; mean read length 9181 bp; maximum read length of 33,305 bp (see Supplementary Fig. S1 for read length distribution). K-mer-based analysis of the PacBio HiFi reads with Genomescope 2.0 estimated a genome size of 443.95 Mb and 0.133 % sequencing error. The k-mer spectrum shows an unimodal distribution with a single major peak at ~64 (Fig. 2A).
Nuclear genome assembly
The size of the final assembly (iyPreImpa1) is similar but not equal to the estimated value from GenomeScope2.0 (Fig. 2A), as has been observed in other taxa (see Pflug et al. (2020) for example). The assembly consists of 787 scaffolds spanning 327.28 Mb with contig N50 of 0.90 Mb, scaffold N50 of 18.68 Mb, longest contig of 12 Mb and largest scaffold of 28.29 Mb. The BUSCO completeness score estimated corresponds to 96.5% using the Hymenoptera gene set, a per base quality (QV) of 56.50, a k-mer completeness of 99.07%, and a frameshift indel QV of 43.84.
During manual curation, we made 359 joins and 24 breaks. In the gap-closing step, we were able to close a total of 8 gaps. We filtered out 2 contigs, 1 corresponding to mitochondrial contamination and 1 corresponding to a mollusc, Ophicardelus ornatus. No other contigs were removed. Assembly statistics are reported in Table 2, and its graphical representation is presented in Fig. 2B. We deposited the genome assembly on GenBank (see Table 2 and Data availability section for details).
Mitochondrial genome assembly
We assembled a mitochondrial genome for P. imparis with MitoHiFi. The final mitochondrial sequence has a size of 18,821 bp, with base composition of A = 44.43%, C = 5.117%, G = 10.32%, T = 40.14%, and consisting of 21 unique transfer RNAs and 12 protein-coding genes. The mitochondrial genome assembly is available on GenBank (see Table 2 and the Data availability section for details).
Assembly comparisons
Genome metrics indicate that the winter ant assembly was highly contiguous (787 scaffolds, scaffold N50 of 18.68 Mb), with a scaffold number and scaffold N50 comparable with other available ant genomes generated with long-read sequencing methods (Fig. 1C, Supplementary Table S1). Although chromosome assignments were not determined for P. imparis, 16 out of the 787 total scaffolds in the genome assembly have sizes >2 Mb (mean ± SD = 17.50 ± 6.35 Mb), make up >85% of the genome assembly, and are comparable with the average chromosome sizes of genome assemblies from 6 representative ant species (mean ± SD = 16.75 ± 8.42 Mb) including the closely related Lasiini species N. fulva and L. platythorax (Fig. 1D, Supplementary Table S2).
Discussion
This genome assembly for the winter ant, P. imparis, adds to a growing number of genomic resources for the diverse and species-rich ant subfamily Formicinae. P. imparis, in particular, is one of the most widespread and recognizable ants in North America, and is particularly abundant during cold seasons, at colder times of the day, and in colder microhabitats. The adaptations that have allowed P. imparis to thrive in these thermal and microgeographic niches also make it especially vulnerable to increasing environmental temperatures, as predicted under future climate change scenarios. Thus, with this new genome assembly, P. imparis is now poised to become a model system for addressing questions related to the genetic basis of thermal adaptation, tolerance of extreme temperatures, and responses to climate change in an important insect clade.
The P. imparis genome assembly presented here has excellent coverage (65.73×) and a high level of BUSCO completeness (96.5%, compared with Hymenoptera, Table 2). Compared with other ant genome assemblies, this winter ant genome is highly contiguous with scaffold and scaffold N50 values similar to that of the chromosome-level genome assemblies of 2 closely related Lasiini species, L. platythorax and N. fulva, as well as other ant species sequenced with long-read methods (Fig. 1C, Supplementary Table S1). The 16 largest P. imparis scaffolds comprise 85.5% of the genome assembly and are similar to the assembled chromosome sizes of L. platythorax (n = 15, Feldmeyer et al. 2023), N. fulva (n = 16, Allen et al. 2023), and 4 representative ant species (Fig. 1D, Supplementary Table S2). Notably, however, the chromosome number suggested by the assembly of California P. imparis (n = 16) is double the previously described chromosome number of Prenolepis nitens collected in Switzerland (2n = 16, n = 8, Hauschteck 1962; note that European P. imparis was revised to P. nitens by [Williams and LaPolla 2016]). It is unclear if this difference represents interspecific karyotypic variation between the European and North American species or misreporting of a haploid specimen from Europe as diploid. Studies of another Prenolepis species, Prenolepis jerdoni, also support a larger haploid chromosome number (n = 16–27, Goni et al. 1982; Imai et al. 1984), which is similar to the chromosome number suggested by the assembly results of California P. imparis. Taken together, these results indicate that this P. imparis genome assembly not only provides a high-quality resource for studying the genetics and evolution of this species but also suggests significant chromosomal variation within the genus, warranting further cytogenetic and genomic investigations.
The assembled size of the P. imparis genome (327.3 Mb) is larger than previous genome size estimates by flow cytometry of samples from Orange County, California (4 individuals; 296.2 ± 2.2 Mb; Tsutsui et al. 2008). Genome assemblies for other ants in the Lasiini tribe include the tawny crazy ant, N. fulva (375.2 Mb, Allen et al. 2023), which has a larger genome than P. imparis, and 3 Lasius species including the jet black ant, Lasius fuliginosus (256.2 Mb, NCBI: GCA_949152495.1), black garden ant, L. niger (236.3 Mb, Koronov et al. 2017), and L. platythorax (235.8 Mb, Feldmeyer et al. 2023) all of which have notably smaller genomes than P. imparis. These differences in genome size highlight the unique genomic characteristics of P. imparis within the Lasiini tribe, underscoring the importance of this new genome assembly for studies of species-specific traits and genome evolution.
The genome assembly of the winter ant, P. imparis, will be a valuable resource for insect conservation, particularly for understanding the response of cold-adapted insects to a warming climate. Moreover, because P. imparis exhibits a continental-scale range, from the Pacific coast to the Atlantic Ocean, it is a particularly appealing model for understanding phylogeography, population differentiation, local adaptation, and dispersal. This genome sequence will also help guide our understanding of Prenolepis populations in California, which include an extremely rare social parasite, will clarify the taxonomy of the species in this genus, and will contribute to larger goals of the CCGP (Shaffer et al. 2022; Toffelmier et al. 2022). Future population genomic studies that include samples from a broader geographic distribution, including a variety of different climates, elevations, and latitudes, will provide insights into the genetic mechanisms underlying thermal adaptation. This information will, in turn, inform future effective conservation and management strategies for this and other species as they contend with a changing climate (Fiedler et al. 2022).
Supplementary material
Supplementary material can be found at http://www.jhered.oxfordjournals.org/.
esae066_suppl_Supplementary_Table_S1
esae066_suppl_Supplementary_Table_S2
esae066_suppl_Supplementary_Figure
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Abdennur N , Mirny LA. Cooler: scalable storage for Hi-C data and other genomically labeled arrays. Bioinformatics.2020:36:311–316. doi: https://doi.org/10.1093/bioinformatics/btz 54031290943 PMC 8205516 · doi ↗ · pubmed ↗
- 2Allen ML , Saelao P, Miles GP, Cross DC, Hill JG, Vargo EL, Grodowitz MJ. Complete chromosome-level genome assembly data from the tawny crazy ant, Nylanderia fulva (Mayr) (Hymenoptera: Formicidae). Data Br 2023:46:108833. doi: https://doi.org/10.1016/j.dib.2022.108833 PMC 980017536591376 · doi ↗ · pubmed ↗
- 3Allio R , Schomaker-Bastos A, Romiguier J, Prosdocimi F, Nabholz B, Delsuc F. Mito Finder: efficient automated large-scale extraction of mitogenomic data in target enrichment phylogenomics. Mol Ecol Resour.2020:20:892–905. doi: https://doi.org/10.1111/1755-0998.1316032243090 PMC 7497042 · doi ↗ · pubmed ↗
- 4Camacho C , Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, Madden TL. BLAST+: architecture and applications. BMC Bioinf.2009:10:421.10.1186/1471-2105-10-421PMC 280385720003500 · doi ↗ · pubmed ↗
- 5Challis R , Richards E, Rajan J, Cochrane G, Blaxter M. Blob Tool Kit – interactive quality assessment of genome assemblies. G 3 Genes|Genomes|Genetics 2020:10:1361–1374. doi: https://doi.org/10.1534/g 3.119.40090832071071 PMC 7144090 · doi ↗ · pubmed ↗
- 6Cheng H , Jarvis ED, Fedrigo O, Koepfli K-P, Urban L, Gemmell NJ, Li H. Haplotype-resolved assembly of diploid genomes without parental data. Nat Biotechnol.2022:40:1332–1335. doi: https://doi.org/10.1038/s 41587-022-01261-x 35332338 PMC 9464699 · doi ↗ · pubmed ↗
- 7Feldmeyer B , Guiglielmoni N, Kirangwa J, Menzel F, Salces-Ortiz J, Fernández R, Atienza EB, Ciofi C, Diroma MA, Iannucci A, et al Reference genome of the ant Lasius platythorax. bio Rxiv 2023:2023. doi: https://doi.org/10.1101/2023.07.18.549505 · doi ↗
- 8Fiedler PL , Erickson B, Esgro M, Gold M, Hull JM, Norris J, Shapiro B, Westphal M, Toffelmier E, Shaffer HB. Seizing the moment: the opportunity and relevance of the California Conservation Genomics Project to state and federal conservation policy. J Hered.2022:113:589–596.36136001 10.1093/jhered/esac 046PMC 9709969 · doi ↗ · pubmed ↗