Sexual epigenetics: genome-wide analysis revealed differential DNA methylation in the vector tick Haemaphysalis longicornis
Han Wang, Ziyan Bing, Lu Li, Ziwen Gao, Chuks Fidelis Nwanade, Na Dong, Ke Li, Leyan Du, Zhijun Yu

TL;DR
This study explores how DNA methylation differs between male and female ticks, providing insights into epigenetic mechanisms behind sex differences in Haemaphysalis longicornis.
Contribution
The study identifies genome-wide DNA methylation differences between male and female ticks, offering new insights into epigenetic regulation of sex-specific traits.
Findings
Female ticks showed higher methylation levels than males, especially in gene body regions.
10,460 differentially methylated regions were identified, with enrichment in binding and metabolic pathways.
Methylation patterns varied by sex and genomic context, suggesting roles in regulating sex-specific functions.
Abstract
Haemaphysalis longicornis is an important vector that transmits a variety of pathogens to humans and animals. This tick species is unique for having two separate reproductive populations: bisexual and parthenogenetic populations. In bisexual populations, morphological differences exist between the males and females, with the females often larger than the males. DNA methylation, as a key epigenetic modification, plays a crucial role in biological processes such as the maintenance of normal cellular function, the regulation of gene expression, and embryonic development. However, the epigenetic mechanism underlying sex differentiation in the bisexual population of H. longicornis has been overlooked. In the present study, the global DNA methylation profiles of the female and male H. longicornis ticks from the bisexual population were explored using whole-genome bisulfite sequencing.…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
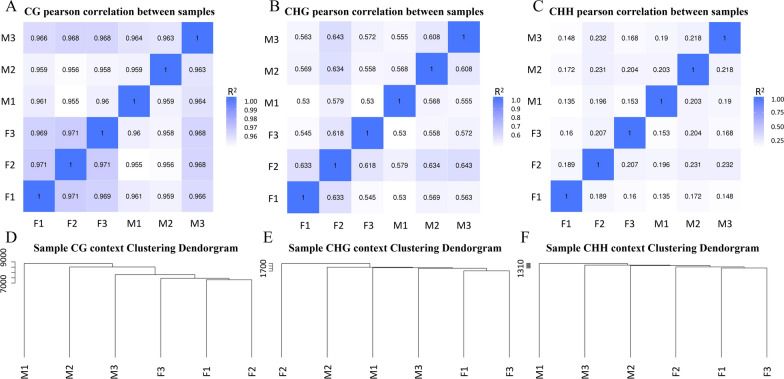 Figure 1
Figure 1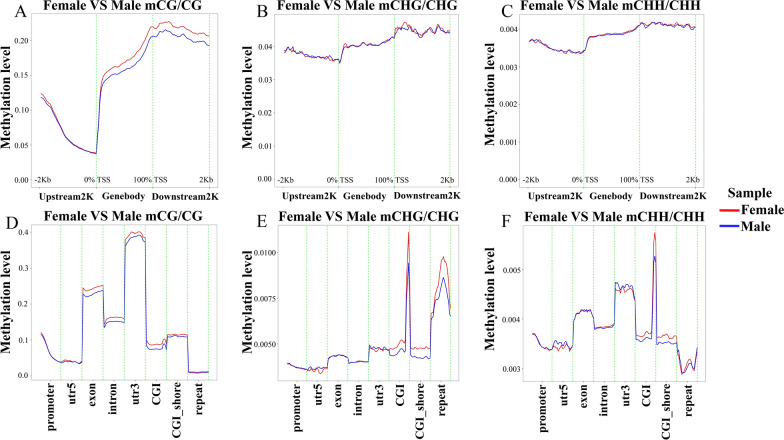 Figure 2
Figure 2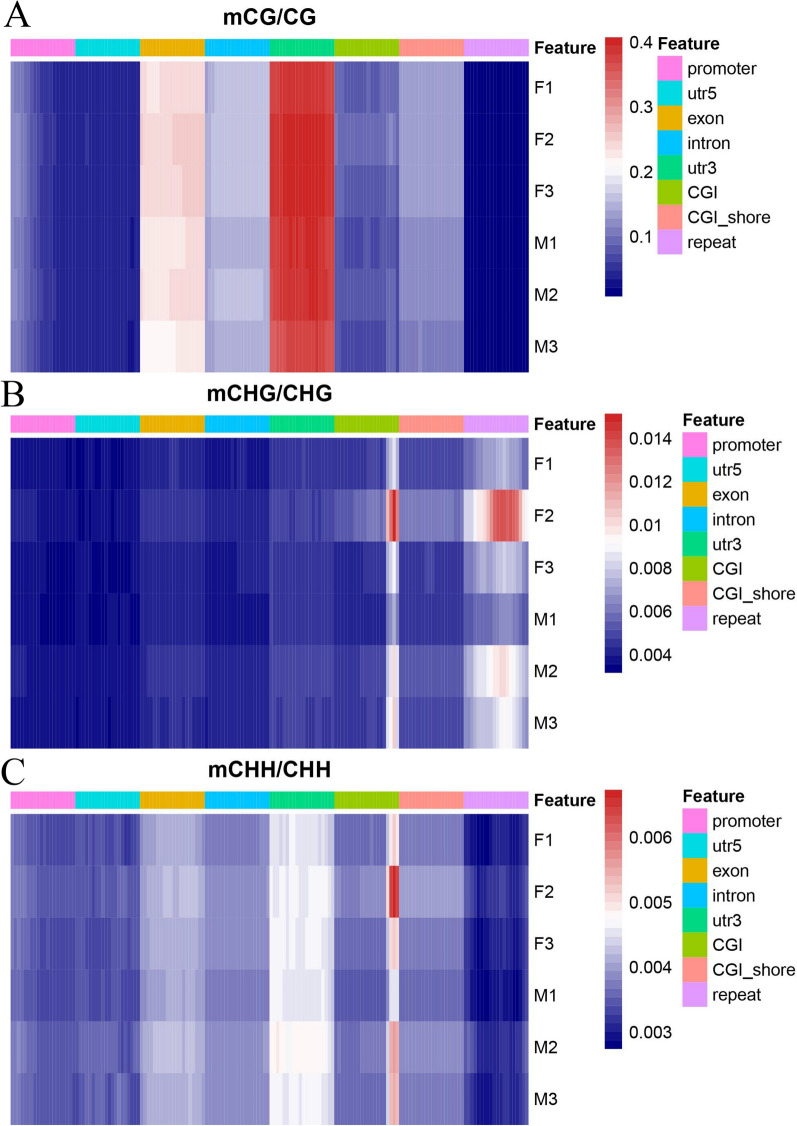 Figure 3
Figure 3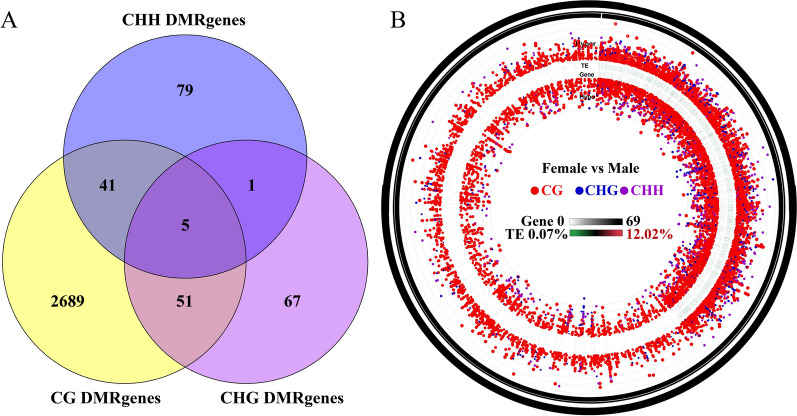 Figure 4
Figure 4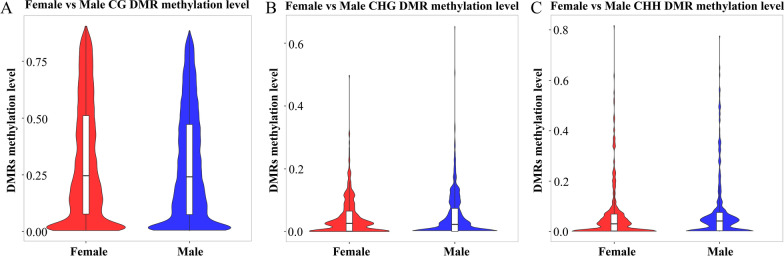 Figure 5
Figure 5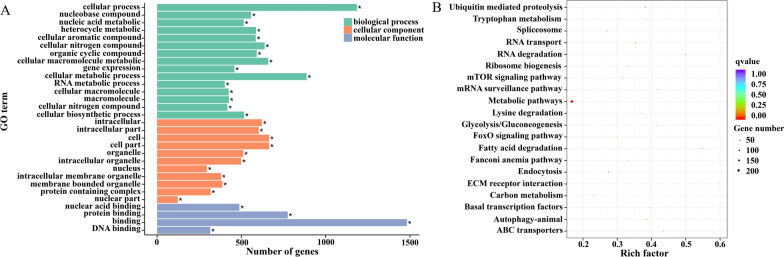 Figure 6
Figure 6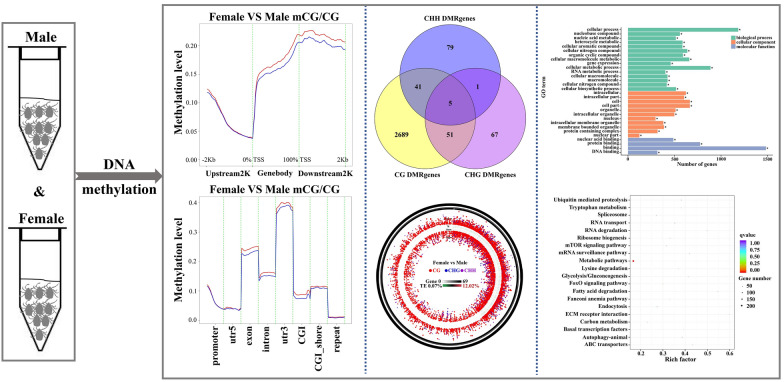 Figure 7
Figure 7- —Funds of Central Guidance for Local Scientific and Technological Development
- —National Natural Science Foundation of China
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsEpigenetics and DNA Methylation · Cancer-related gene regulation
Background
Sexual dimorphism is a common phenomenon across a wide range of sexually reproducing organisms. It is often considered a diverse feature and refers to the behavioral, morphological (physical), and physiological differences between males and females [1, 2]. While some of these differences are driven by genes situated on the sex chromosomes [3], others have been associated with other genomic processes, such as epigenetic mechanisms and alternative splicing [4, 5]. These mechanisms contribute to the phenomenon known as sex-biased gene expression, which is widely recognized as a factor influencing most of the sex-specific differentiation [2].
DNA methylation is a crucial epigenetic modification involved in diverse biological processes [6]. Major forms include 5-methylcytosine (5mC) and N^6^-methyladenine (m^6^A) [7]. DNA methylation in insects occurs in the transcriptional regions of genes; the levels are generally lower than in vertebrates [8] and show great variation between arthropods [9]. DNA methyltransferases (DNMTs) catalyze the formation of 5mC by transferring methyl groups from S-adenosylmethionine (SAM) to the fifth carbon of cytosine residues [10]. DNA methylation regulates gene expression in both eukaryotes and prokaryotes and is involved in various biological processes including development, nutrigenomics, tumorigenesis, and DNA repair [10–12]. Despite some evidence suggesting the possible involvement of DNA methylation in sexual differentiation across various arthropods [13–15], differential DNA methylation patterns remain unclear in ticks. Nevertheless, recent research has found DNA methylation patterns in ticks in response to cold tolerance [7]. To advance our understanding in this area, additional research is needed to explore methylation patterns of male and female ticks. This is of significant interest since it can provide insights into the biological mechanisms behind tick sexual differences (sexual dimorphism). By manipulating these epigenetic processes, new approaches to controlling ticks and associated pathogens can potentially be developed.
Among globally distributed tick species, Haemaphysalis longicornis is native to East Asia but is considered an invasive species in regions such as Australia, New Zealand, and the United States [16, 17]. This tick consists of a diploid bisexual population, a triploid parthenogenetic population, and an aneuploid population capable of parthenogenetic and bisexual reproduction [18]. The parthenogenetic populations are found in Australia, New Zealand, Korea, Japan, the United States, and some areas in China (Sichuan and Shanghai), whereas the bisexual populations are widely distributed across China [19, 20]. The female H. longicornis can ingest a great amount of blood, whereas the males feed less and display remarkable morphological differences from females. This tick has been associated with a variety of human and animal pathogens and can transmit bacteria, fungi, viruses, and parasites [21]. These pathogens cause disease in humans, wildlife, and livestock, making them a potential threat to public and veterinary health [22]. Given their long life cycles and unique ecological adaptations, ticks may have evolved a highly complex set of epigenetic regulatory mechanisms. This can provide a strong adaptive response and buffer capacity in the changing natural environment, thus effectively ensuring the sustainability of its survival and reproduction process [23].
In this study, the DNA methylation status of adult males and females of H. longicornis was comprehensively analyzed using the whole-genome bisulfite sequencing (WGBS) technique, and sex specific DNA methylation profiles were assessed. These findings provide new insights into the epigenetic regulation of sex development in H. longicornis, and further expand our understanding of reproductive divergence and epigenetics in H. longicornis.
Methods
Ticks
Unfed adults of H. longicornis were collected from vegetation by flagging/dragging at the Xiaowutai Mountain National Nature Reserve Area, Hebei Province, China. For feeding, ticks were put into cloth bags attached to the ears of domestic rabbits [24]. Second-generation adults (20 females and 20 males) 2 weeks post-molting were randomly selected for the subsequent experiment, which was carried out three times. Cytological studies have found that the number of chromosomes of the bisexual tick was 21–22 [19], and from this, combined with stereoscopic and scanning electron microscopy observation of the morphology of female genital pores and Haller's organ [18], it was determined that the ticks used represented a bisexual population. During the unfed period, they were placed in an incubator (26 ± 1 °C, 75 ± 5% relative humidity [RH], 16:8 h [light/dark, L:D] photoperiod). All experiments involving rabbits were approved by the Animal Ethics Committee of Hebei Normal University (Protocol Number: IACUC-209230).
DNA quantification and qualification
Genomic DNA was extracted using a Magnetic Universal Genomic DNA Kit (Tiangen Corporation, Beijing, China), following the manufacturer's recommendations. Genomic DNA degradation and contamination were validated by agarose gels. DNA purity was checked using a NanoPhotometer^®^ spectrophotometer (Implen, CA, USA). DNA concentration was measured using a Qubit^®^ DNA Assay Kit in a Qubit^®^ 2.0 Fluorometer (Life Technologies, CA, USA). DNA quality assessment data for the WGBS are listed in Table S1.
Library preparation and quantification
A total of 100 ng genomic DNA spiked with 0.5 ng lambda DNA was fragmented by sonication to 200–300 base pairs (bp) with the Covaris S220 Focused-ultrasonicator. These DNA fragments were treated with bisulfite using the EZ DNA Methylation-Gold™ Kit (Zymo Research), and the library was constructed by Novogene Corporation (Beijing, China). Subsequently, paired-end sequencing of the sample was performed on the Illumina platform (Illumina, CA, USA). Library quality was assessed on an Agilent 2100 Bioanalyzer system.
Data analysis
The library was sequenced on the Illumina NovaSeq platform. Image analysis and base calling were performed with the Illumina CASAVA pipeline, and finally, 150-bp paired-end reads were generated. The sequenced raw data have been deposited in the National Center for Biotechnology Information (NCBI) Sequence Read Archive (SRA) with accession number PRJNA938509.
Quality control
First, basic statistics on the quality of the raw reads were performed using FastQC (fastqc_v0.11.5). Then, those read sequences produced by the Illumina pipeline in FASTQ format were pre-processed through fastp (fastp 0.20.0). The remaining reads that passed all the filtering steps were counted as clean reads, and all subsequent analyses were based on this. Finally, we used FastQC to perform basic statistics on the quality of the clean data reads.
Reference data preparation before analysis
Prior to the analysis, the reference data for the species under study were prepared, which included the reference sequence in FASTA format, the annotation file in gtf format, the GO annotation file, the description file, and the gene region file in bed format. Repeats were predicted using RepeatMasker, and the CpG-island (CGI) track was generated from a genome using cpgIslandExt.
Read mapping to the reference genome
Bismark software (version 0.16.3) was used to perform alignments of bisulfite-treated reads to a reference genome (–X 700 dovetail) [25]. The reference genome was first transformed into a bisulfite-converted version (C-to-T and G-to-A conversion) and then indexed using bowtie2 [26]. Clean reads were also transformed into fully bisulfite-converted versions (C-to-T and G-to-A conversion) before being aligned to the similarly converted versions of the genome in a directional manner. Sequence reads that produced a unique best alignment from the two alignment processes (original top and bottom strand) were then compared to the normal genomic sequence, and the methylation state of all cytosine positions was inferred. Reads that aligned to the same regions of the genome were regarded as duplicate reads. The sequencing depth and coverage were summarized using de-duplicated reads.
The results of the methylation extractor (bismark_methylation_extractor, – no_overlap) were transformed into bigWig format for visualization using the Integrative Genomics Viewer (IGV) browser. The sodium bisulfite non-conversion rate was calculated as the percentage of cytosine sequenced at cytosine reference positions in the lambda genome.
Estimating methylation level
Methylated sites were identified with a binomial test using the methylated counts (mC), total counts (mC + unmethylated count [umC]), and the non-conversion rate (r). Sites with a false discovery rate (FDR)-corrected P-value < 0.05 were considered as a methylated site. To calculate the methylation level of the sequence, we divided the sequence into multiple bins, with a bin size of 10 kilobases (kb). The sum of methylated and unmethylated read counts in each window was calculated. The methylation level for each window or C site shows the fraction of mCs, and is defined as follows:
\documentclass[12pt]{minimal} \usepackage{amsmath} \usepackage{wasysym} \usepackage{amsfonts} \usepackage{amssymb} \usepackage{amsbsy} \usepackage{mathrsfs} \usepackage{upgreek} \setlength{\oddsidemargin}{-69pt} \begin{document}$$ {\text{ML}}\left( {\text{C}} \right) \, = {\text{reads}}\left( {{\text{mC}}} \right)/{\text{reads}}\left( {{\text{mC}}} \right) \, + {\text{ reads}}\left( {\text{C}} \right), $$\end{document}where ML is the methylation level.
Differential methylation analysis
Differentially methylated regions (DMRs) were identified using Decision Support System (DSS) software [27–29]. The core of DSS is a new dispersion shrinkage method for estimating the dispersion parameter from gamma-Poisson or beta-binomial distributions. According to the distribution of DMRs through the genome, we defined the genes related to DMRs as genes whose gene body region (from transcription start site [TSS] to transcription end site [TES]) or promoter region (2 kb upstream from the TSS) overlapped with the DMRs.
Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analysis of DMR-related genes
GO enrichment analysis of genes related to DMRs was implemented using the GOseq R package [30], in which gene length bias was corrected. GO terms with corrected P-values less than 0.05 were considered significantly enriched by DMR-related genes. The KEGG Orthology-Based Annotation System (KOBAS) was utilized to assess the statistical enrichment of genes associated with DMRs within KEGG pathways [31].
Results
Whole-genome bisulfite sequencing and DNA methylation
In this study, to investigate the role of DNA methylation in the sexual dimorphism development of H. longicornis, WGBS technology was applied to sequence three repetitive sequences of female and male adult H. longicornis. During the sequencing process, female groups generated an average of 316,263,220 raw reads, while male groups generated 326,746,634. After quality control and filtering, approximately 309,318,166 reads in the female groups and 317,373,434 reads in the male groups were recognized as pure reads. Of these, 40.46% of the female sample reads and 39.44% of the male sample reads were uniquely mapped to the reference genome of the ticks, showing high sequencing quality and data accuracy. Repeat rates were similar between groups: 24.65% (females) and 24.57% (males). In addition, the bisulfite conversion rate exceeded 99.68% for both groups, which further demonstrated the high confidence of the WGBS data (Table 1).Table 1. Summary of bisulfite (BS) sequencing of the tick Haemaphysalis longicornisFemaleMaleRaw reads316,263,220326,746,634Raw bases (G)94.8898.02Clean reads309,318,166317,373,434Clean bases (G)84.6586.39Clean ratio (%)89.2288.14Q20 (%)97.1096.67Q30 (%)91.0990.19GC content (%)24.2024.14BS conversion rate (%)99.6899.68Mapped reads125,144,776125,038,818Mapping rate (%)40.4639.44Duplicate rate (%)24.6524.57
In the analysis of DNA methylation levels, the proportion of methylated cytosines in the three sequence environments, CG, CHG, and CHH, was of particular interest. In female ticks, approximately 0.56% of the genomic C site was methylated, while in males, this proportion was slightly lower at 0.47%. In the CG sequence environment, the methylation level was 2.30% in females and 1.92% in males. It may be that more repetitive raw reads in the male sample were filtered, and fewer methylation sites were ultimately retained. In the CHG and CHH sequence environments, the methylation levels were almost the same between the two groups, 0.04% and 0.01%, respectively (Table 2).Table 2. Proportion of methylated C sites occurring in different sequence environmentsMethylated CMethylated C percentagemC in CGMethylated CG percentagemC in CHGMethylated CHG percentagemC in CHHMethylated CHH percentageFemale8,414,0410.56%8,150,9052.30%127,0710.04%136,0660.01%Male6,997,4830.47%6,744,3651.92%124,8350.04%128,2870.01%
Sample correlation and cluster analysis
The correlation of methylation levels between groups was analyzed as a key indicator for assessing the reliability of the study. Pearson correlation analyses were performed on duplicate groups of female and male H. longicornis based on data in the CG, CHG, and CHH sequence environments. In the CG sequence environment, the Pearson correlation coefficients between both female and male groups exceeded 0.95, a result that indicates a strong correlation between the two groups of groups (Fig. 1A). In the CHG sequence environment, the Pearson correlation coefficients between the two groups of groups ranged from 0.5 to 0.7, which indicated a weak correlation between the two groups of groups (Fig. 1B). In contrast, the Pearson correlation coefficients of the two groups of groups in the CHH sequence environment ranged from 0.1 to 0.3, which indicated a lack of correlation between the two groups of groups (Fig. 1C).Fig. 1. Pearson correlation based on the CG/CHG/CHH context among the replicate groups. A Pearson correlation based on CG context among the replicate groups. B Pearson correlation based on CHG context among the replicate groups. C Pearson correlation based on CHH context among the replicate groups. D Sample CG context clustering dendrogram. E Sample CHG context clustering dendrogram. F Sample CHH context clustering dendrogram. R^2^ Pearson's correlation coefficient; F female group; M male group
When the groups were analyzed for clustering based on CG methylation patterns, males and females clustered separately (Fig. 1D). In the CHG sequence environment, a certain degree of clustering tendency could still be observed (Fig. 1E). Males and females also clustered separately in the CHH sequence environment (Fig. 1F).
DNA methylation levels in functional regions
Differences in methylation levels were assessed in three sequence environments, CG, CHG, and CHH, as well as in different gene regions (DMRs). The results showed that within the 2-kb region upstream of the TSS, the differences in methylation levels between the two groups were not significant. However, in the gene body region and downstream 2-kb region, the methylation level of female groups was slightly higher than that of male groups, and the peak methylation appeared in the downstream region near the TES (Fig. 2A). Figure 2B and C demonstrate that there was little difference in methylation levels between the sexes in the CHG and CHH sequence environments. In the differential analysis of methylation levels in different gene regions, the 3′ untranslated region (UTR) had the highest methylation level in the CG sequence environment, followed by exon, intron, and CGI_shore, and the methylation levels of female groups were generally higher than those of male groups. In contrast, the methylation levels of the 5′ UTR and repeat regions were relatively low (Fig. 2D). In the CHG and CHH sequence environments, the highest methylation levels were found in the CGI region and were higher in female groups than in male groups. In the repeat region, the CHG type had a higher methylation level, while the CHH type had the lowest methylation level, a finding that is reflected in Fig. 2E and F. To demonstrate these differences more visually, heat maps were used to show the differences in methylation levels in CG, CHG, and CHH sequence environments in DMRs (Fig. 3).Fig. 2. Distribution of methylation levels of functional regions, and the gene upstream and downstream between the female group and male group. A Distribution of methylation level of functional regions between female group and male group in mCG/CG. B Distribution of methylation level of functional regions between female group and male group in mCHG/CHG. C Distribution of methylation level of functional regions between female group and male group in mCHH/CHH. D Distribution of methylation level of genes upstream and downstream between female group and male group in mCG/CG. E Distribution of methylation level of genes upstream and downstream between female group and male group in mCHG/CHG. F Distribution of methylation level of genes upstream and downstream between female group and male group in mCHH/CHH. TSS transcription start site, TES transcription end siteFig. 3Heat map of differences in methylation levels in different gene regions. A Heat map of differences in methylation levels in mCG/CG. B Heat map of differences in methylation levels in mCHG/CHG. C Heat map of differences in methylation levels in mCHH/CHH
Analysis of DMRs
To further elucidate the role of DNA methylation in the sexual dimorphism development of H. longicornis, an analysis was conducted to identify DMRs and differentially methylated genes (DMGs) in adult females and males. A total of 10,460 DMRs were identified in female and male groups (Table S2). These DMRs were further classified into 5282 hypermethylated DMRs and 5178 hypomethylated DMRs, and the specific information is listed in Tables S3 and S4, respectively.
The DMGs in the context of CG, CHG, and CHH are demonstrated by Venn diagrams. The analysis revealed that 2786, 124, and 126 DMGs were observed in CG, CHG, and CHH sequence environments, respectively, with five DMGs exhibiting differential methylation in all three sequence environments (Fig. 4A). Furthermore, the number of red dots indicates that the most methylation occurs at the CG locus between females and males, while noting that the expression levels of transposons ranged from 0.07% to 12.02% (Fig. 4B). In addition, a violin plot analysis of methylation levels in the DMRs was performed. The results showed that the methylation level was highest in the CG sequence environment. The median value of the DMR methylation level associated with the female groups was slightly higher than that of male groups in the CG and CHH sequence environments, but lower than that of the male group in the CHG (Fig. 5).Fig. 4. The methylation patterns between the female group and male group of H. longicornis. A Venn diagrams of female and male group DMGs under CG, CHG, and CHH contexts of the H. longicornis adults. B Significantly different methylation patterns between the female group and male groupFig. 5Violin plot analysis of the methylation level of DMRs in the female and male groups of H. longicornis adults. A Violin plot analysis of CG DMR methylation level. B Violin plot analysis of CHG DMR methylation level. C Violin plot analysis of CHH DMR methylation level
GO and KEGG pathway enrichment analysis
GO and KEGG analyses were performed to investigate DNA methylation changes in H. longicornis across sexes. Given that the majority of DMGs are located in CG methylation backgrounds, DMG functional enrichment analyses were performed based on CG methylation. GO enrichment analysis was classified into three main categories: biological processes, cellular components, and molecular functions. The results of the analyses showed that biological processes were mainly enriched in cellular processes. The cellular components were mainly enriched in the cell, cell part, and intracellular region, while the molecular functions were primarily enriched in binding (Fig. 6A). In addition, KEGG pathway enrichment analysis was performed on DMGs. The results showed that KEGG pathways were mainly enriched in metabolic pathways (Fig. 6B). These findings help to reveal the biological significance and potential molecular mechanisms of DNA methylation changes in H. longicornis in different sexes.Fig. 6GO and KEGG enrichment analyses of the female and male group CG DMGs of the H. longicornis adults. A GO enrichment analysis of DMGs. B KEGG pathway enrichment
Discussion
Modifications to DNA and histones affect changes in chromatin and the expression of genes that regulate many cellular and developmental processes. DNA methylation plays an important role in regulating a variety of biological processes, such as social insect behavior and population differentiation [32]. Preliminary studies on honeybees suggest that DNA methylation is instrumental in regulating population differentiation and memory processing behavior [33, 34]. Knockdown of the DNA methyltransferase 3 (Dnmt3) gene in honeybees resulted in reduced genome-wide methylation levels and altered patterns of gene RNA splicing, suggesting that Dnmt3 and DNA methylation play an important role in queen differentiation [35]. Studies have shown that king and worker larvae have significantly different methylation patterns, demonstrating that DNA methylation is important in termite Zootermopsis nevadensis grade differentiation [36]. DMGs between populations have been identified in the reproductive and sterile bumblebee workers [37]. In the ants Camponotus floridanus and Harpegnathos saltator, there is a link between DNA methylation and RNA splicing [38]. Dnmt methyltransferases involved in the de novo methylation of tick DNA have been identified in previous studies [39, 40]. However, the role of DNA methylation in the sexual dimorphism development and physiological regulation of the tick has not been clearly defined. The two breeding populations of H. longicornis, the bisexual population and the parthenogenetic population, have different DNA methylation transcription patterns at different life stages [41]. In this study, WGBS was used for the first time to explore DNA methylation changes in a bisexual population of H. longicornis. This allowed us to comprehensively study genome-wide DNA methylation profiles in the bisexual population.
The results of the present study showed that the methylated C sites of H. longicornis female ticks were slightly higher than in male ticks. According to existing research, there are differences in DNA methylation based on sex. While some studies have reported higher methylation levels in females [42], others have found higher methylation levels in males [5, 43] or similar methylation between males and females [2]. These contradictory results across various studies may be attributed to many factors, including the techniques used for DNA methylation analysis, sample size, and the parts or tissues analyzed, indicating the complexity of sex-specific DNA methylation. The slightly higher methylation of the female tick may correlate with females typically becoming larger (engorged) than males after a blood meal, which is essential for egg production and development. In addition, we observed that the methylated genomic C site in the males (0.47%) and females (0.56%) of H. longicornis was lower when compared with a previous study of adult females, which found levels of 0.95% and 0.94% in the control and low-temperature-treated groups, respectively [7]. This demonstrates the importance of exploring DNA methylation in individual species, since methylation can vary within species and/or between sexes, and many factors, such as cell or tissue types, age, and environmental conditions, can result in varying degrees of methylation. Our results also implied that methylation (methylated genomic C site) in ticks, including H. longicornis, is low (less than 1%), although much higher than that in some insects [7].
Based on the sequence of cytosines (CG, CHG, and CHH), we found that the methylation levels in the CG sequence environment were higher in females than males, and levels were almost the same between the two groups in the CHG and CHH sequence environments. This suggests that the different methylation levels across these sequences may play a role in sex-specific differences and that high CG methylation may be associated with sex differences, contributing to the biological characteristics between H. longicornis males and females at the molecular level. Males and females of other arthropods are known to have differential and/or variable CpG methylation patterns [43, 44], which is a potential mechanism underlying sex-specific differences. Our results also suggest that methylation in ticks, including H. longicornis, occurs at the CG site, which is consistent with a previous DNA methylation study of H. longicornis exposed to low-temperature stress [7]. Additionally, our cluster analysis revealed that male and female groups of the studied ticks clustered separately, which was more pronounced in CG and CHH sequence environments, possibly indicating where each group exhibits a specific methylation profile. These results highlight a promising direction for future studies.
By analyzing the DNA methylation levels in different genomic functional regions, we found that the differences showed sex-specific and region/site dependence. For instance, the 3′ UTR region had the highest methylation level in the CG sequence environment, followed by the exon, intron, and CGI_shore regions, suggesting that these regions may have important roles in the regulation of gene expression. It has also been shown that H. longicornis has different functional genomic regions at the CG site, and DNA methylation is more likely to occur in the 3’ UTR region [7]. Moreover, the female groups generally had higher methylation levels than the male groups, especially in the gene body region and downstream regions near the TES, which may affect gene expression and function. Indeed, DNA methylation within the gene body region usually results in higher levels of gene expression, but methylation in other regions/sites may have the opposite effect [45–47]. This situation is often referred to as the “DNA methylation paradox,” where methylation in different sites/regions has opposite effects on a gene, and the specific location of methylation can determine whether the gene is silenced or actively transcribed [47]. Overall, the DNA methylation patterns differed slightly between female and male ticks. Our results suggest that CG methylation may play an important role in the gene regulation of H. longicornis, particularly in a sex-specific context, acting as a key epigenetic mechanism that determines which genes may be expressed and/or repressed in the males and females of this tick species. However, how the different genomic regions or sites (sequence environments) contribute to gene regulation remains unclear, making further exploration necessary.
To further elucidate the role of DNA methylation in the sexual differentiation of H. longicornis, analysis was conducted to identify DMRs and DMGs in adult female and male groups. Overall, we found differentially hypermethylated and hypomethylated regions between the two groups, and a greater number of genes were differentially methylated at the CG, which further demonstrates differential methylation based on sex and regions/sites (sequence environments). DMGs in H. longicornis are involved in several biological processes, cellular components, and molecular functions, and DMGs are significantly enriched in binding and RNA transport during cold stress [7]. Here, our GO and KEGG pathway analyses revealed that DMGs were most significantly enriched in binding and metabolic pathways, suggesting their potential involvement in regulating relevant genes in the males and females of H. longicornis. However, additional studies are needed to gain a comprehensive understanding of their contributions to the regulation of relevant genes and to examine the expression pattern of genes related to males and females of H. longicornis, which will provide exciting new directions for further investigation.
Conclusions
DNA methylation profiles of adult male and female H. longicornis were comprehensively analyzed. Different methylation patterns were found between the sexes, which may affect gene expression related to sexual development. Sex-linked DMRs and DMGs were also identified. Before any conclusion can be drawn regarding the findings of this study, it is imperative to refine the mechanisms and functions of DNA methylation and its broader implications in the sexual differentiation of ticks, including H. longicornis. Nevertheless, these results may provide new clues and targets for the analysis of the mechanism of DNA methylation in the regulation of sex differences and the identification of key genes in ticks.
Supplementary Information
Additional file 1.Additional file 2.Additional file 3.Additional file 4.