DNA sequencing of whole human cytomegalovirus genomes from formalin-fixed, paraffin-embedded tissues from congenital cytomegalovirus disease cases
Kathy K. Li, Nicolás M. Suárez, Salvatore Camiolo, Andrew J. Davison, Richard J. Orton, Michael Nevels, Michael Nevels, Michael Nevels

TL;DR
Researchers successfully sequenced the full genome of a virus causing congenital disease from preserved tissue samples, opening new possibilities for studying how genetic differences affect disease outcomes.
Contribution
Demonstrated feasibility of sequencing whole HCMV genomes from FFPE tissues, expanding sample availability for genetic studies.
Findings
Whole HCMV genomes were successfully sequenced from five cases using FFPE material.
Two commercial DNA extraction kits were evaluated for FFPE HCMV sequencing.
The study provides a pipeline for genome assembly and variant calling from FFPE samples.
Abstract
Congenital cytomegalovirus disease (cCMV) is uncommon but can be severe. Investigations of the role of genome sequence variation in the causative virus (human cytomegalovirus, HCMV) in clinical outcome have to date depended on small sample numbers derived from fresh tissues. Extensive formalin-fixed, paraffin-embedded (FFPE) cCMV biorepositories established worldwide potentially provide much larger sample numbers for future investigations. However, there are no published reports of sequencing whole HCMV genomes from such material. To sequence whole HCMV genomes from cCMV FFPE material Sixteen FFPE samples of foetal kidney or placental tissue were processed from ten cCMV cases in foetuses or neonates. Two commercial kits for extracting DNA from FFPE material were evaluated, HCMV DNA was enriched in the extracts, and the samples were sequenced on the Illumina platform. The sequence read…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
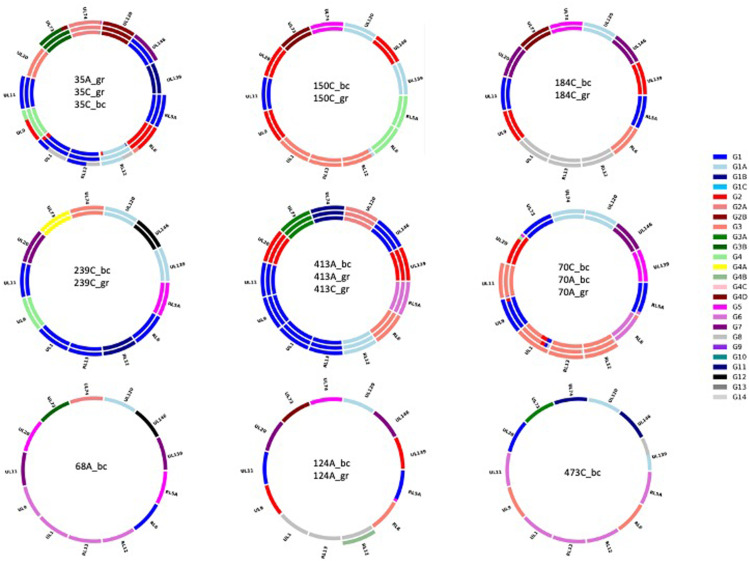 Fig. 1
Fig. 1- —http://dx.doi.org/10.13039/501100000265Medical Research Council
- —http://dx.doi.org/10.13039/501100000265Medical Research Council
- —http://dx.doi.org/10.13039/100010269Wellcome Trust
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsCytomegalovirus and herpesvirus research · Biosensors and Analytical Detection · Herpesvirus Infections and Treatments
Introduction
Congenital cytomegalovirus disease (cCMV) is the most common non-genetic cause of sensorineural hearing loss and neurodevelopmental delay [1]. The role of variation in the causative virus (human cytomegalovirus, HCMV) in clinical outcome has been investigated in several studies [2]. These studies focused on hypervariable HCMV genes in order to determine whether particular genotypes are associated with virulence in single-strain infections, and whether multiple-strain infections are more virulent than single-strain ones. However, as cCMV affects only 1 in 100–150 live births [3], access to clinical samples is limited. Biorepositories of formalin-fixed, paraffin-embedded (FFPE) tissues commonly collected in pathology departments thus offer a resource for future studies.
Archived placental FFPE samples have proved useful as an adjunct in diagnosing infants asymptomatic of cCMV at birth, and some studies have used such samples to detect HCMV by immunohistochemistry or PCR amplification of short genomic fragments [4,5]. However, to our knowledge, no published work has involved sequencing whole HCMV genomes from FFPE material. This is due largely to the difficulty of recovering DNA of sufficient quality [6], as formalin adversely affects nucleic acid integrity.
Objective
To assess the feasibility of sequencing whole HCMV genomes from archived FFPE material.
Materials and methods
Sixteen FFPE samples of placental or foetal kidney tissue from ten cCMV cases (2008–2018) were retrieved from the pathology archive at Birmingham Women’s Hospital, UK. The associated pseudonymised data were collected by a member of the primary care team on 18 September 2018. These samples, labelled with delinked reference numbers, were sent with the pseudonymised data to the MRC-University of Glasgow Centre for Virus Research for sequencing. Ethical approval was granted by the Health Research Authority Research Ethics Committee (HRA REC reference 18/LO/1441; R&D number 18/BW/NNU/NO17; 31 August 2018), and consent for future research on excess samples was obtained at the time of sampling by the primary care team for tissues retained in the Birmingham biorepository. The authors had no access to patient-identifiable data during or after the study. The cases included five from intra-uterine death, two from termination of pregnancy, one from miscarriage, and two from neonatal death (Table 1).
Table 1: Pseudonymised metadata from cCMV cases used in this study.
Two kits for extracting DNA from FFPE material via different methodologies were assessed: one using a paramagnetic bead-based approach (FormaPure DNA extraction and purification kit, Beckman Coulter) and the other using spin-column technology (GeneRead DNA FFPE kit, QIAGEN). DNA load in the extracted samples was determined using a Qubit fluorometer (ThermoFisher Scientific), and HCMV and human DNA loads were determined by qPCR targeting the HCMV UL97 [7] and human FOXP2 genes [8], respectively (S1 Table). Only samples with an HCMV load >100 IU/μL were processed for sequencing. The extracts were enriched for HCMV DNA by hybridisation-based capture [9] and sequenced on the Illumina platform. GRACy, a software pipeline for determining HCMV genome sequences from Illumina data [10], was used to analyse each sequence read dataset by read filtering, genotyping, genome assembly and variant (single nucleotide polymorphism; SNP) calling.
The read filtering module removed human reads, trimmed adapters and low-quality nucleotides, and removed duplicate reads.
The genotyping module enumerated sequence motifs in the filtered datasets that were specific to the genotypes of 13 hypervariable HCMV genes, thus allowing the number of HCMV strains in a sample to be estimated without requiring genome assembly. For each dataset, a more stringent threshold than that used for fresh clinical samples, akin to that used in human genetics for FFPE samples, was applied to assign genotypes to each gene: > 100 reads representing >5% of reads detected for all genotypes of that gene [11,12,13,14]. The number of strains was then registered as being the greatest number of genotypes detected for at least two genes, with a requirement for consistent assignment of genotypes across datasets from the same case. In addition, this module determined whether the combination of 13 genotypes for each dataset was represented among a large collection of published HCMV genome sequences.
The genome assembly module produced a draft HCMV sequence from each dataset. The original datasets for each case were then combined, processed using Trim Galore v.0.4.0 (https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/), and aligned to the best draft assembly for that case using Bowtie 2 v2.4.2 [15] with the --local parameter. The read alignment was visualised using Tablet v1.21.02.08 [16], and improvements were implemented manually to yield the final sequence. Read coverage was determined by aligning each dataset to the final sequence. The variant calling module applied a threshold similar to that used commonly in human somatic allelic calling: a frequency of 5% [11,14] and a coverage of 50 reads/nt.
Results
DNA extracts of sufficient quality for sequencing were obtained from all cases but case 660 (S1 Table). These included 11 extracts from nine cases using the FormaPure kit and eight extracts from six cases using the GeneRead kit. Extracts prepared using the GeneRead kit contained more DNA but had higher A260/280 ratios (indicative of residual RNA) than those prepared using the Formapure kit (S1 Fig.). However, there was no significant difference between the two kits in the quality of the HCMV sequence data generated, as assessed from the average coverage depth of a reference HCMV genome (S1 Fig.).
Genotyping was carried out for 19 datasets from 12 FFPE samples from nine cCMV cases (Fig 1). Analysis of three datasets (124R_fp, 35R_gr and 70P_fp) did not meet threshold requirements probably because of a combination of low DNA load and low proportion of HCMV DNA (S1 Table). Analysis of the remaining 16 datasets indicated that eight cases involved a single HCMV strain and one (case 70) may have involved one or more additional minor strains. None of the combinations of 13 genotypes for each dataset was represented among published HCMV genome sequences. This is consistent with prior evidence that, due to intrastrain recombination during HCMV evolution, vast numbers of genotype combinations exist among natural strains [12,17,18].
Doughnut plots reporting HCMV genotypes from dataset analysis.Each ring represents an individual dataset, and is divided into sections representing the 13 hypervariable genes analysed. Datasets are listed from the outer ring inwards. The size of the coloured bars corresponds to the proportion of genotypes detected for each gene, as coded in the panel on the right using published genotype nomenclature (https://github.com/salvocamiolo/minion_Genotyper/blob/master/depositedSequences_codes.txt). Blank segments indicate that genotyping failed thresholds. Dataset names consist of the case number suffixed by P (placenta) or R (kidney) and then by _fp (FormaPure extraction kit) or _gr (GeneRead extraction kit).
Whole genome sequences were determined for five cases (Table 2) with relatively high HCMV load. The sequences from cases 413 and 239 exhibit unusual characteristics. The HCMV genome (236 kbp) has the structure ab-U_L_-b’a’c’-U_S_-ca, where U_L_ and U_S_ are long and short unique regions, respectively, flanked by inverted repeats a, b and c and their reverse complements a’, b’ and c’. For case 413, two versions (318 and 288 bp) of a subsequence of c/c’ were detected in approximately equal proportions. These versions may be present in a single genome population with one subsequence in c and the other in c’, or they may be segregated into two populations with identical copies in c and c’ in each. For case 239, the a sequence at the left genome end differs from the a’ sequence internally, the latter consisting of two fused, dissimilar a’ sequences and the former being identical to one of these sequences except for 8 bp at one end. These characteristics were present in both the placental and kidney samples from each case and were therefore unlikely to have been artefactual.
Table 2: Coverage statistics and deposition data for read datasets and genome sequences.
Variant calling identified 14 SNPs distributed among four cases (Table 3). All but one SNP was present in a single dataset at low frequency, and ten were C:G to T:A mutations, which occur in FFPE samples due to hydrolytic deamination of C residues to form U residues. Seven of the C:G to T:A mutations were detected in samples extracted using the FormaPure kit, which, unlike the GeneRead kit, does not incorporate uracil-DNA glycosylase to remove mismatched U residues. A single SNP was detected in both samples from case 239 at high frequency (≥36%).
Table 3: SNPs detected at levels over the threshold.
Discussion
This study met its objective by demonstrating that whole HCMV genomes may be sequenced from cCMV FFPE material. This was achieved with samples that had been archived for up to five years; it is possible that low HCMV load, rather than poor quality DNA, was the main contributor to low read coverage in older samples. Given the scarcity of fresh cCMV samples and the consequent small number and geographical restrictions of samples employed in published studies on the role of HCMV variation and strain composition in clinical outcome [2], this advance may result in FFPE repositories located worldwide proving key to future studies.
Ancillary data on the number of HCMV strains in the samples (by genotyping) and the occurrence of SNPs (by variant calling) were also obtained in this study, but, given the limitations mentioned above, conclusions relating to clinical outcome were not an objective. Future work would profit not only from the greater sample numbers that FFPE repositories afford but also from investigating additional steps for preserving or repairing DNA integrity in FFPE material, with the objective of reducing the effects of formalin-induced artefacts on variant calling, and from side-by-side comparisons with fresh cCMV material.
Supporting information
S1 TableCharacteristics of extracts used to generate sequence datasets.(DOCX)
S1 FigPlots characterising FFPE extracts prepared using the FormaPure or GeneRead kits and sequence data generated from these extracts.(DOCX)
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Manicklal S, Emery VC, Lazzarotto T, Boppana SB, Gupta RK. The “silent” global burden of congenital cytomegalovirus. Clin Microbiol Rev. 2013;26(1):86–102. doi: 10.1128/CMR.00062-12 23297260 PMC 3553672 · doi ↗ · pubmed ↗
- 2Arav-Boger R. Strain variation and disease severity in congenital cytomegalovirus infection: in search of a viral marker. Infect Dis Clin North Am. 2015;29(3):401–14. doi: 10.1016/j.idc.2015.05.009 26154664 PMC 4552582 · doi ↗ · pubmed ↗
- 3Dollard SC, Grosse SD, Ross DS. New estimates of the prevalence of neurological and sensory sequelae and mortality associated with congenital cytomegalovirus infection. Rev Med Virol. 2007;17(5):355–63. doi: 10.1002/rmv.544 17542052 · doi ↗ · pubmed ↗
- 4Folkins AK, Chisholm KM, Guo FP, Mc Dowell M, Aziz N, Pinsky BA. Diagnosis of congenital CMV using PCR performed on formalin-fixed, paraffin-embedded placental tissue. Am J Surg Pathol. 2013;37(9):1413–20. doi: 10.1097/PAS.0b 013e 318290 f 171 23797721 · doi ↗ · pubmed ↗
- 5de la Cruz-de la Cruz A, Moreno-Verduzco ER, Martínez-Alarcón O, González-Alvarez DL, Valdespino-Vázquez MY, Helguera-Repetto A-C, et al. Utility of two DNA extraction methods using formalin-fixed paraffin-embedded tissues in identifying congenital cytomegalovirus infection by polymerase chain reaction. Diagn Microbiol Infect Dis. 2020;97(4):115075. doi: 10.1016/j.diagmicrobio.2020.115075 32534239 · doi ↗ · pubmed ↗
- 6Gilbert MTP, Haselkorn T, Bunce M, Sanchez JJ, Lucas SB, Jewell LD, et al. The isolation of nucleic acids from fixed, paraffin-embedded tissues-which methods are useful when?. P Lo S One. 2007;2(6):e 537. doi: 10.1371/journal.pone.0000537 17579711 PMC 1888728 · doi ↗ · pubmed ↗
- 7Slavov SN, Otaguiri KK, de Figueiredo GG, Yamamoto AY, Mussi-Pinhata MM, Kashima S, et al. Development and optimization of a sensitive Taq Man® real-time PCR with synthetic homologous extrinsic control for quantitation of Human cytomegalovirus viral load. J Med Virol. 2016;88(9):1604–12. doi: 10.1002/jmv.24499 26890091 · doi ↗ · pubmed ↗
- 8Soejima M, Hiroshige K, Yoshimoto J, Koda Y. Selective quantification of human DNA by real-time PCR of FOXP 2. Forensic Sci Int Genet. 2012;6(4):447–51. doi: 10.1016/j.fsigen.2011.09.006 22001153 · doi ↗ · pubmed ↗