A novel c.116 - 117 del variant in Unverricht-Lundborg disease: first ULD report in large Chinese population and review of the pathogenetic variants in CSTB gene
Pu Miao, Yao Ding, Zhidong Cen, Yulan Chen, Wei Luo, Baorong Zhang, Zhiying Wu, Meiping Ding, Shuang Wang

TL;DR
This paper reports the first case of Unverricht-Lundborg disease in a large Chinese population and identifies a new genetic variant linked to the condition.
Contribution
The study presents the first ULD case in China and identifies a novel c.116 - 117 delAG variant in the CSTB gene.
Findings
The patient had a novel indel variant (c.116 - 117 delAG) and a CSTB gene repeat expansion.
Perampanel treatment effectively reduced seizures and myoclonic jerks in the patient.
Compound or homozygous point/indel variants were associated with earlier symptom onset and more refractory seizures.
Abstract
Unverricht-Lundborg disease (ULD) is a rare autosomal recessive neurodegenerative disorder, often caused by biallelic promoter expansions of CSTB gene or, more rarely by point/indel variants. The best-known area for ULD are the shores of the Baltic and Mediterranean Sea and few cases have been recorded from Asia. In this report, we present the first case of a Chinese patient with ULD. The patient was a 21-year-old female with normal cognitive function. She developed nocturnal bilateral tonic–clonic seizures (BTCS) at age 8, with subsequent onset of myoclonic jerks along with ataxia at age 12. Myoclonic jerks were triggered by flashing lights and during menstrual periods. EEG recording showed multifocal spikes and sharp-waves, predominantly in bilateral occipital regions. Genetic testing revealed heterozygous compound variants for a novel indel variant (c.116 - 117 delAG) and the repeat…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
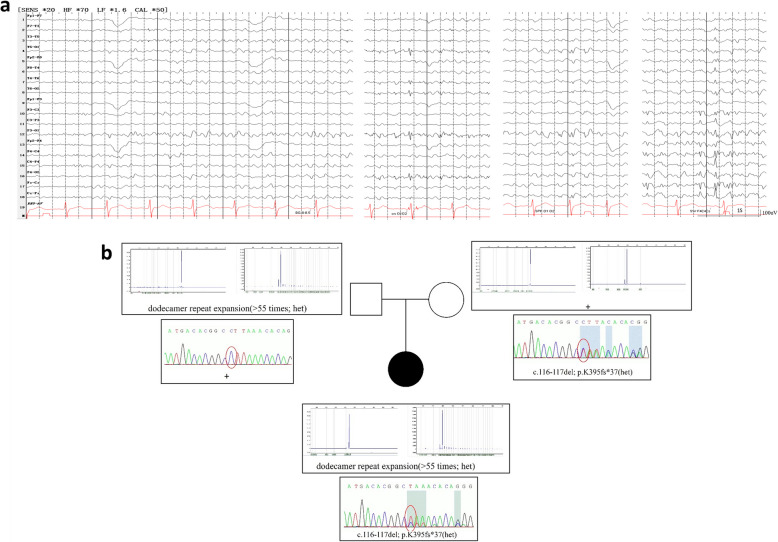 Figure 1
Figure 1- —http://dx.doi.org/10.13039/100014717National Outstanding Youth Science Fund Project of National Natural Science Foundation of China
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsGlycogen Storage Diseases and Myoclonus · Lysosomal Storage Disorders Research · Genetics and Neurodevelopmental Disorders
Background
Unverricht-Lundborg disease (ULD) is an autosomal recessive neurodegenerative disorder caused by biallelic alterations of the CSTB gene. ULD is characterized by bilateral tonic–clonic seizures (BTCS), myoclonus, mild cognitive decline, and other neurological symptoms such as ataxia, dysarthria, and psychiatric disorders [1]. The most frequent genetic cause of ULD is the unstable expansion (> 30 times) of a dodecamer repeat expansion. In rare cases it can also occur in compound heterozygous (a dodecamer repeat expansion and a sequence variant such as single-nucleotide variant or indel), or even in homozygous CSTB point or indel variants [2, 3]. To date, less than 20 pathogenic CSTB disease-causing variants have been reported in ULD patients [4]. Therefore, profound relationship between genotype and phenotype has not been fully established.
ULD is most reported in Finland, where the total ULD population is estimated to be only around two hundreds [5]. While sporadic cases have been reported globally, including in Asian countries such as Japan and Korea, no cases of ULD have been documented in the Chinese population, despite it being the world's largest ethnic group, with over 1.4 billion people worldwide. It is worth noting that about 10% of ULD patients exhibit a mild phenotype, which could result in underdiagnosis [5]. However, it could not explain why such a large population has never been documented to have ULD patients.
Here, we report a case of ULD with a definite genetic diagnosis in a Chinese patient. Our patient was found to have a compound heterozygous variant of a novel indel (c.116 - 117 delAG) and a dodecamer repeat expansion in the CSTB gene, confirming the existence of the pathogenic genetic background of CSTB gene in the Chinese population. We also reviewed and compared the phenotype of patients with different genetic types of CSTB gene.
Case presentation
The patient was a 21-year-old female, graduating from the junior high school. She was born at term by non-consanguineous parents and her psychomotor development was normal. She reported no family history of epilepsy or other neurological disorders. Her first bilateral tonic–clonic seizure (BTCS) occurred when she was 8 years old and was triggered by bright lights. She subsequently experienced recurrent nocturnal convulsive seizures. Initially, she was treated with oxcarbazepine, which proved ineffective. At the age of 12, she developed myoclonic jerks that were usually triggered by flashing lights and menstrual periods. Valproate was introduced, which reduced the frequency of myoclonic jerks. However, her symptoms worsened at the age of 18, and both valproate and levetiracetam showed a poor response. She experienced 1–2 BTCSs per month (all occurring at night), 2–3 myoclonic seizures per week, and myoclonic jerks triggered by light or when she was in a state of tension.
The patient was referred to our hospital for preoperative evaluation of refractory epilepsy at the age of 19. The neurological examination revealed no abnormalities except for truncal ataxia. Her brain MRI scan was normal. FDG-PET revealed hypometabolism especially affecting the bilateral frontal-parietal junction regions. Video-EEG recordings showed multifocal spike-wave and sharp-wave epileptiform discharges, predominantly in bilateral occipital regions (electrodes O_1_ and O_2_, Fig. 1a), with normal posterior background during interictal periods. After perampanel was introduced at 2 mg daily, she had a marked improvement in myoclonic jerks and gait. Epileptic seizures, including BTCS and myoclonic seizures, stopped with the use of perampanel (PER). During the two-year follow-up, she remained seizure-free but experienced recurrent symptoms of myoclonic jerks, which responded to PER dose escalation (up to 6 mg/day). No psychological or behavioral side-effects of PER were observed during the follow-up. The patient is currently taking perampanel 6 mg/day and levetiracetam 1000 mg/day.Fig. 1a EEG recording showed normal background and multifocal spike-wave, sharp-wave epileptiform discharges, predominantly in bilateral occipital regions (O_1_, O_2_ electrodes). b Genetic findings of the patient. Pedigree of the family and PCR amplification-capillary electrophoresis and sanger sequencing showing the segregation of the variants in the family members
A PCR-amplification test of the CSTB gene identified a pathogenic heterozygous dodecamer repeat expansion (≥ 55 times). Combining this with the clinical phenotype, it supported a diagnosis of ULD disease and necessitated the identification of another variant. Whole-exome sequencing revealed a previously unpublished heterozygous variant (c.116 - 117 del, p.K395fs* 37) in the CSTB gene (Fig. 1b). According to the American College of Medical Genetics and Genomics (ACMG) guidelines, it could be evaluated as a likely pathogenic variant (PVS1 + PM2). Further verification confirmed it as a compound heterozygosity.
Discussion
We have presented the first genetic confirmed case of ULD in the Chinese population, identified through mutations the CSTB gene. Our patient had a ten-year misdiagnosis, highlighting the under-reporting of ULD in China. ULD, a form of progressive myoclonic epilepsy (PME), can be difficult to distinguish from juvenile myoclonic epilepsy (JME) and familial cortical myoclonic tremor with epilepsy (FCMTE) in the early stages of the disease. However, the active myoclonus features and the responses to antiseizure medications (ASMs) can be helpful for early identification. Conventional PCR may not be effective in detecting the size of fully expanded alleles and GC-rich sequences in the dodecamer repeat region, which likely contributes to the underdiagnosis of ULD in China. Additionally, the insufficient application of this special genetic testing in China further compounds this issue.
Another noteworthy aspect of ULD is the genotype–phenotype correlation. The CSTB gene, the primary genetic causes of ULD, is situated on chromosome 21 at the 21q22.3 locus. This relatively small gene comprises three exons and spans approximately 3 kilobases (kb) of genomic DNA [6]. It encodes the Cystatin B protein, a member of the cysteine protease inhibitor family, which is also referred to as Stefin B. The CSTB gene is widely expressed in neural cells and plays a role in brain development such as cell proliferation differentiation and neuronal migration [7]. The CSTB gene is the primary genetic cause of ULD and includes three pathogenic types of variants: 1) homozygous promoter expansions in the CSTB gene (observed in approximately 80–90% of probands), 2) compound heterozygosity for a CSTB dodecamer repeat expansion and a CSTB sequence variant, and 3) homozygosity for a CSTB sequence variant. In our case, compound heterozygosity with a novel variant, c.116 - 117 delAG (deletion of two nucleotides in exon 2), was found. This variant is predicted to result in a truncated CSTB protein due to a frameshift (p.K39SfsX37), which might lead to a loss of function of the CSTB protein. To date, 13 pathogenic variants in 24 patients, including descriptions of the phenotype, have been reported in association with ULD. By comparing with a group of ULD patients with homozygous promoter expansions in the CSTB gene (mean age of first seizures:10.0 ± 2.28 years, range: 5–13 years old, n = 16) [1], patients with compound heterozygosity or homozygosity for a CSTB sequence variant (mean age of first seizures: 7.5 ± 3.00 years, range: 0.5–13 years, n = 16) presented with an earlier age of onset (Mann–Whitney test, P = 0.013), as previously reported by Canafoglia et al. [2]. An earlier onset has been associated with a worse prognosis; therefore, the disease course in patients with a pathogenic CSTB sequence variant may be less favorable than in patients with common homozygous promoter expansions.
In the majority of cases in Table 1 (78.5%, 11/14), BTCS was recorded as the first symptom, and this percentage was higher than in patients with homozygous promoter expansions [8]. It has been reported that BTCS has a positive response to antiseizure medications (ASMs) and may disappear as the disease progresses. However, our patient suffered from BTCS for nearly ten years and had a poor response to escalating dose of valproic acid (VPA) and levetiracetam (LEV). Similar findings were reported by Koskenkorva et al. among patients with the c.202 C > T variant [7]. None of the patients in Table 1 were on monotherapy, and a few patients even required more than four ASMs. BTCS among patients with compound heterozygosity or homozygosity for a CSTB sequence variant might become refractory to ASMs more easily than in patients with homozygous promoter expansions. The earliest and most used medication was VPA, but nearly all patients had a poor response. Our patient suffered from a long-standing nocturnal BTCS, and a novel medication, PER, that selectively inhibits postsynaptic AMPA receptors, demonstrated a remarkable effect. Although an abatement of PER efficiency for her myoclonic jerks was observed, her BTCS remained well-controlled during the two-year follow-up. PER may be recommended as an effective early treatment option for these patients, especially those with refractory BTCS. Table 1. Description of main clinical and genetic features in ULD patients harboring point or indel disease causing variants of the CSTB geneNumY/GAge at onsetSeizures (year)TreatmentCognitive impairmentAtaxiaMood disorder/walking abilityEEGBrain MRICSTB variants NM_000100.4TypesDescent1 [8]/6–16BTCS often presenting as the first symptom; stimulus-sensitive myoclonusSW-PSW/218_219 delTC, CFrame shiftFrance2 [8]169 - 2 A > G, CSplice siteFrance3 [8]10G > C, HMissenseMorocco4 [3]18, F0.5BTCS (0.5)/SevereYesNone/Wheelchair-boundSW+ Cerebral atrophy218 dupT, HFrame shiftSri Lankan5 [3]10, FNoneNoneNoneSevereYesNone/Wheelchair-boundDiffuse slowingCerebral atrophySri Lankan6 [9]33, M14BTCS (14), M(18)PHT, VPA, CZP, LEV, ZNS+VNSNormalYesNone/NormalSW-PSW; IPS-MNormalc.66G > A, HSplice sitePortugal7 [10] > 19, M/Myoclonic (/)//Yes//Abnormalc.10G > T, HMissenseEastern8 [2]42, M7M (7), BTCS (10)VPA, LEV, ZNS, ESM, CZPModerateYesYes/-SW; IPS-MNormalc.67 - 1G > C, CSplice siteItalian9 [2]35, M9BTCS (9), M (15)VPA, LEV, CZP, PRMModerateYesNone/-SW+ Normalc.67 - 1G > C, CSplice siteSpanish10 [2]34, M5BTCS (5), M(7)VPA, LEV, ESM, CZPSevereYesNone/-SW-PSW; IPS-MCerebral atrophyc.67 - 1G > C, CSplice siteSpanish11 [2]30, M6BTCS (6), M(16)VPA, LEV, LTG, CZPNormalYesNone/-SW+;IPS-MNormalc.168 + 1_18 del, CFrame shiftItalian12 [2]23, M9BTCS (9), M(12)VPA, LEV, LTG, DZPMildNoneNone/-PSWNormalc.133 C > T, CMissenseItalian13 [2]15, M6BTCS (6), M(8)ZSM, VPA, CZPMildYesYes/-SW-PSW; IPS-MAbnormalc.168 + 2–168 + 21 delinsA, CSplice siteItalian14 [2]16, M7BTCS (7), M (9)ZNS, VPA, LEV, CZP, PIRMildYesYes/-SW-PSW; IPS-MNormalItalian15 [2]12, M10BTCS (10), M (10)LEV, PERNormalNoneNone/-SW-PSW; IPS-MNormalItalian16 [11]18, M8M (8), BTCS(13)VPA, CBZ, CZP, ZNP, PIRMildNoneNone/-SW; IPS-MNormalc.168G > A, CSplice siteJapanese17 [12]49, M8M (8), BTCS (13)VPA, LEV, PIR, TPM, CZPMildYesYes/-SW+ Normalc.132_134 delGAG, CIn frameRoman18 [12]53, F13BTCS (13), M(13)VPA, PHTNormalNoneYes/-SWNormalRoman19 [7]23, M5BTCS, MVPA, LTG, LEV, PIRSevereNoneNone/Limited walking abilitySW+; IPS-MCerebral atrophyc.202 C > T, CNonsenseFinnish20 [7]21, M7BTCS,MVPA, LTG, LEVMildNoneSW+; IPS-MNormalFinnish21 [7]34, F10BTCS, MVPA, CZP, TPM, PIRModerateNoneNone/Wheelchair boundSW+; IPS-MCerebellar atrophyFinnish22 [7]37, M7BTCS, MVPA, CZP, LEV, PIRModerateNoneSW+; IPS-MBrain atrophyFinnish23 [7]14, F6BTCS, MVPA, CZPSevereYesNone/NormalSW+; IPS-MNormalFinnish2621, F8BTCS (8),M (12)OXC, VPA, LEV, PERNormalYesYes/NormalSW; IPS-MNormalc.116 - 117 del, CDeletionChineseF Female, M Male, BTCS Bilateral tonic–clonic seizure, M Myoclonous, SW Spike weave, SW*+* Spike weave with diffuse background slowing, IPS-M IPS-induced myoclonus, CH Compound heterozygous, Hom Homozygous, VPA Valproate, LEV Levetiracetam, ZNS Zonisamide, ESM Ethosuximide, CZP Clonazepam, LTG Lamotrigine, PIR Piracetam, PER Perampanel, CBZ Carbamazepine, TPM Topiramate, PHT Phenytoin
In addition, only a fraction of the pathogenic variants are missense (3/13, 23%). Interestingly, we found that patients with missense variants might exhibit a milder phenotype. It has been reported that homozygous promoter expansions leads to a reduced expression of the CSTB gene, and lower levels of CSTB expression are associated with a more severe phenotype [3, 4]. Variants affecting splice sites or predicting truncated protein may have a greater impact on CSTB expression than missense variants, which could potentially explain the varying degrees of phenotype severity. Further investigations should focus on understanding the mechanism by which reduced CSTB expression leads to distinct phenotypes.
Conclusions
In conclusion, we report the first case of ULD in the Chinese population with an unpublished variant c.116 - 117 del in the CSTB gene. When combined with other reported compound or homozygous variants, patients with this variant manifested an earlier onset and more refractory BTCS than those with homozygous promoter expansions in the CSTB gene. Among ULD patients with compound or homozygous variants, missense variants account for only a small percentage and their phenotype may be milder.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Lucchino V, Scaramuzzino L, Scalise S, Lo Conte M, Zannino C, Benedetto GL, et al. Insights into the Genetic Profile of Two Siblings Affected by Unverricht-Lundborg Disease Using Patient-Derived hi PS Cs. Cells. 2022;11(21):3491.10.3390/cells 11213491 PMC 965599236359887 · doi ↗ · pubmed ↗
- 2Koskenkorva P, Hyppönen J, Aikiä M, Mervaala E, Kiviranta T, Eriksson K, et al. Severer phenotype in Unverricht-Lundborg disease (EPM 1) patients compound heterozygous for the dodecamer repeat expansion and the c.202C>T mutation in the CSTB gene. Neurodegener Dis. 2011;8(6):515–22.10.1159/00032347021757863 · doi ↗ · pubmed ↗
- 3Lalioti MD, Mirotsou M, Buresi C, Peitsch MC, Rossier C, Ouazzani R, et al. Identifcation of mutations in cystatin B, the gene responsible for the Unverricht-Lundborg type of progressive myoclonus epilepsy (EPM 1). Am J Hum Genet. 1997;60(2):342–51.PMC 17123899012407 · pubmed ↗
- 4Matthews AM, Blydt-Hansen I, Al-Jabri B, Andersen J, Tarailo-Graovac M, Price M, et al. Atypical cerebral palsy: genomics analysis enables precision medicine. Genet Med. 2019;21(7):1621–8.10.1038/s 41436-018-0376-y 30542205 · doi ↗ · pubmed ↗
- 5Kagitani-Shimono K, Imai K, Okamoto N, Ono J, Okada S. Unverricht-Lundborg disease with cystatin B gene abnormalities. Pediatr Neurol. 2002;26(1):55–60.10.1016/s 0887-8994(01)00336-811814737 · doi ↗ · pubmed ↗
- 6Assenza G, Benvenga A, Gennaro E, Tombini M, Campana C, Assenza F, et al. A novel c 132-134del mutation in Unverricht-Lundborg disease and the review of literature of heterozygous compound patients. Epilepsia. 2017;58(2):e 31–e 35.10.1111/epi.1362627888502 · doi ↗ · pubmed ↗