A 43-year-old female with personality changes and alien limb
Anza Zahid, Zbigniew K Wszolek, Abdul R Alchaki

TL;DR
A 43-year-old woman with personality changes and alien limb symptoms is reported, highlighting a rare CSF1R-related disorder.
Contribution
This paper presents a case report and clinical discussion of a rare CSF1R-related disorder with underdiagnosed symptoms.
Findings
CSF1R-related disorder can present with neurocognitive decline and alien limb phenomenon.
The case highlights the importance of considering CSF1R mutations in differential diagnosis.
Treatment options and clinical approaches for managing the disorder are discussed.
Abstract
Colony stimulating factor-1 receptor (CSF1R) encodes for a tyrosine kinase receptor expressed on microglia. CSF1R related disorder is a devastating autosomal dominant leukoencephalopathy caused by CSF1R with variable penetrance in adults. It remains significantly underdiagnosed or misdiagnosed. We report a case of a 43-year-old woman with an insidious onset of neurocognitive decline, alien limb phenomenon, and personality changes over 1 year. In this report we will discuss the clinical approach, differential diagnosis, investigation, and available treatment options for CSF1R related disorder.
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
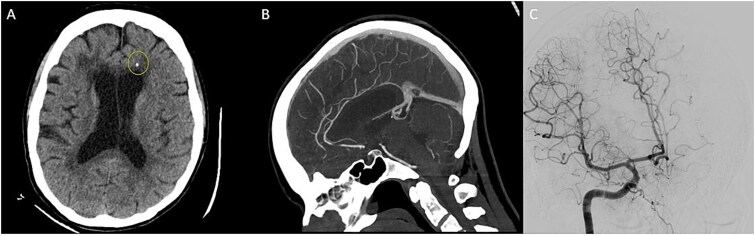 Figure 1
Figure 1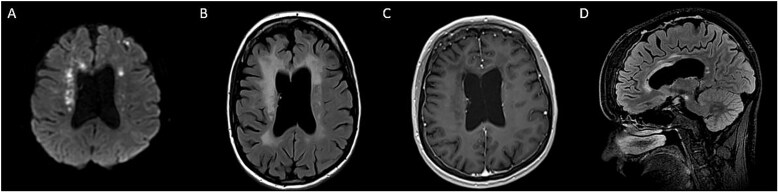 Figure 2
Figure 2Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsNeuroinflammation and Neurodegeneration Mechanisms · Alzheimer's disease research and treatments · Tryptophan and brain disorders
Case report
A 43-year-old right-handed woman presented with left extremity loss of function and a change in personality over 1 year at the neurology outpatient clinic. After the birth of her fourth child, she began to experience frequent headaches. She also became withdrawn, forgetful, and volatile in her mood—crying often and laughing inappropriately. She did not have a family history of neuropsychiatric or memory disorders.
On the MoCA, she scored 13 points out of 30, losing points in visual–spatial testing, delayed memory, and calculation. Neurological exam revealed oculomotor apraxia, 3 beats of nystagmus on horizontal right-sided gaze. Grasp reflex was present bilaterally. She demonstrated significant in-coordination of her left hand, stating ‘her left hand has a mind of it’s own’. She had increased tone in all her extremities with an admixture of rigidity and spasticity on the left side but her. Motor strength was normal. Deep tendon reflexes were exaggerated bilaterally. Sensory testing was normal. On gait assessment, she walked unassisted dragging of her left foot in planter flexed position. Romberg’s test was negative.
Head CT showed ventriculomegaly and punctate calcification in the frontal lobe (Fig. 1A). Brain MRI showed diffusion restriction, T2 periventricular hyperintensity with atrophy of the genu and anterior body of the corpus callosum (Fig. 2). Cerebrospinal Fluid (CSF) revealed WBC 2/CMM, RBC 435/CMM, Protein 48 mg/dL, Glucose 62 mg/dL (serum glucose 80 mg/dl), a normal IgG index and synthetic rate.
CT head showing punctate calcification in the frontal lobe (A). CT angiogram without evidence of occlusion or venous thrombosis (B). Cerebral angiogram of R carotid artery was unremarkable without evidence of beading (C).
Brain MRI showing diffusion restriction (A) with T2 FLAIR confluent periventricular hyperintensity, ventricular enlargement due to frontoparietal atrophy (B) and atrophy of genu and anterior body of the corpus callosum (D) without enhancement (C).
Based on patients’ history of headaches, left-sided weakness, cognitive and personality changes differential diagnosis include cerebral venous sinus thrombosis, vascular etiology (stroke, Cerebral Autosomal Dominant Arteriopathy with Subcortical Infarcts and Leukoencephalopathy (CADASIL), Susac’s disease), neurodegenerative conditions such as behavioral variant of Fronto-temporal dementia, Corticobasal degeneration, post-COVID demyelination syndrome, or multiple sclerosis.
Frontal brain biopsy revealed extensive myelin loss with macrophage infiltration and presence of neuroaxonal spheroids. The genetic testing confirmed the diagnosis that was positive for heterozygous CSF1R c.1765G > A, (p.Gly589Arg) gene mutation. No pathogenic variant for NOTCH3 gene was detected. Therefore, a diagnosis of CSF1R-related disorder was confirmed. (Dulski J, et al Parkinsonism Related Disorders, 2024).
She was given a trial of high dose steroids without significant improvement. Induction dose of intravenous immunoglobulin (2 g/kg) showed transient benefit per oral report. For left upper extremity spasticity she was offered botulinum toxin injections.
Discussion
Genetic testing was positive for a heterozygous mutation in CSF1R c.1765G > A, (p.Gly589Arg) that confirmed the diagnosis for *CSF1R-*related disorder.
CSF1R-related disorder is an autosomal dominant leukoencephalopathy with variable penetrance. One third to half cases are sporadic due to novel mutation and genetic mosaicism [1]. CSF1R encodes colony stimulating factor 1 receptor, a tyrosine kinase receptor expressed on microglia that was first identified in a cluster of families presenting with adult-onset leukoencephalopathy. Almost all mutations are located in the tyrosine kinase domain (TKD) of the CSF1R. Thus, the activation of CSF1R through autophosphorylation is required for signal transductions, which contributes to microglial maintenance and activation [2]. Advances in molecular genetics and the discovery of causative genes have facilitated increasing recognition of the disease.
The mean age at onset of CSF1R-related disorder is 43 years [3]. Women are observed to develop the disease 7 years earlier than men [3]. As observed in our patient, the disease is clinically characterized neuropsychiatric symptoms and parkinsonism [2].
Typical radiographic findings in CSF1R-related disorder includes scattered subcortical and periventricular calcifications with ‘stapping stone’ appearance on Head CT, patchy to confluent white matter abnormalities in the frontal and parietal lobes without gadolinium enhancement (Fig. 2) [3]. U-fibers are usually preserved. Thinning of corpus callosum with enlargement of ventricle and persistent diffusion restriction. [4] Subcortical high DWI signals in *CSF1R-*related strongly corelate with pathological spongiotic changes, primary axonal degeneration and loss of myelin sheaths [5]. The persistence is also hypothesized to reflect persistent intramyelinic edema in regions of neurodegeneration [5, 6]. Our patients imaging did not reveal classic radiographic features observed in CSF1R related disorders. She only had one small calcification without ‘stapping stone’ appearance of calcification in classic cases. Thus, brain biopsy was considered.
Historically, these patients have been misdiagnosed with multiple sclerosis or demyelinating disease, Cerebral Autosomal Dominant Arteriopathy with Subcortical Infarcts and Leukoencephalopathy (CADASIL), primary angiitis, Alzheimer Disease and fronto-temporal dementia. [3, 7] However, varying clinical and radiographic features can help differentiate CSF1R-related disorders from other disorders.
The neuropsychiatric symptoms manifest as a progressive decline in memory, depression, apathy, irritability, and behavior changes are observed in behavioral variant frontotemporal dementia (bvFTD). The imaging of our patient did not demonstrate typical fronto-temporal atrophy to be suggestive of bvFTD. Similarly, CADASIL can present with symptoms of unilateral weakness, dementia, migraine headaches. The classic imaging findings in CADASIL include hyperintense white matter signal abnormalities in the anterior temporal poles, centrum semi-ovale, external capsule, basal ganglia and pons along with evidence for acute infarcts and micro-hemorrhages. It is associated with a genetic mutation in NOTCH3 gene that was negative in our patient. Cerebral angiogram was unremarkable for findings suggestive of a primary angiitis.
Similarly, both CSF1R- and *AARS2-*related leukoencephalopathy share several neurological symptoms and can present with similar white matter involvement, predominantly in the frontoparietal and periventricular regions [8]. However, the differences in radiological images between the two gene encoding mutations have been identified in the corpus collosum, in the regions with severe brain atrophy and in patients with AARS2 gene mutations lack the unique calcifications that are seen on the computed tomography (CT). Unlike CSF1R, the AARS2-related phenotypes are also observed in adolescence [8].
Pathology may support the diagnosis with the presence of spheroid and pigmented glia, however, there have been reports of identical pathology in *AARS2-*related disorder without the pathogenic mutation. Hence, genetic testing is the most specific test to confirm the diagnosis [2]. (https://doi.org/10.5061/dryad.498j63f).
Unfortunately, there is no cure for CFS1R-related disorder. Steroids, cyclophosphamide, interferons B1a/1b, and plasmapheresis are the mainstay of treatment [9]. Symptomatic treatment and multidisciplinary approach with anti-depressants, antipsychotics, botulinum toxin injections for spasticity, rehabilitation, and referral to genetic counselling are offered. [2]
Allogenic hematopoietic stems cell transplantation (HSCT) from Human Leukocyte Antigen (HLA) matched wild-type CSF1R donors may offer a potential regenerative strategy in slowing neurological progression and extending survival of the patients to about 15 years in selected cases [10]. Acute or chronic graft versus host disease was not observed with HSCT, however, worsening neurological symptoms including; extrapyramidal symptoms, parkinsonism, pneumonia, and new localization-related seizures has been reported [10].
IGNITE Phase 2 trial of Iluzanebart (VGL101) using TREM2 agonist is showing promise at 6-month assessment in patients with CSF1R-related disorder. Iluzanebart (VGL101) is a humanized monoclonal antibody that binds with TREM2 to activate the signaling pathway to increase the ability of microglia and to protect the neurons from damage (NCT05677659). [1, 2]
Early genetic testing is critical in patients with possible CSF1R related disorder to enable early treatment and slow disease progression [2].
Patient outcome
Our patient’s disease had progressed too far, rendering her ineligible for clinical trials. She was given a trial of high dose steroids without significant improvement. Induction dose of intravenous immunoglobulin (2 g/kg) showed transient benefit per oral report. For left upper extremity spasticity she was offered botulinum toxin injections. The family was referred to a genetic clinic for counseling for her 4 children.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Papapetropoulos S, Gelfand JM, Konno T. et al. Clinical presentation and diagnosis of adult-onset leukoencephalopathy with axonal spheroids and pigmented glia: a literature analysis of case studies. Front Neurol 2024;15:1. 10.3389/fneur.2024.1320663 PMC 1096238938529036 · doi ↗ · pubmed ↗
- 2Konno T, Kasanuki K, Ikeuchi T. et al. CSF 1R -related leukoencephalopathy. Neurology 2018;91:1092–104. 10.1212/WNL.000000000000664230429277 PMC 6329328 · doi ↗ · pubmed ↗
- 3Konno T, Yoshida K, Mizuno T. et al. Clinical and genetic characterization of adult-onset leukoencephalopathy with axonal spheroids and pigmented glia associated with <scp>CSF</scp> 1R mutation. Eur J Neurol 2017;24:37–45. 10.1111/ene.1312527680516 PMC 5215554 · doi ↗ · pubmed ↗
- 4Konno T, Broderick DF, Mezaki N. et al. Diagnostic value of brain calcifications in adult-onset leukoencephalopathy with axonal spheroids and pigmented glia. Am J Neuroradiol 2017;38:77–83. 10.3174/ajnr.A 493827633805 PMC 5233547 · doi ↗ · pubmed ↗
- 5Huang H, Cao L, Chen H. Dynamic analysis of CSF 1R-related leukoencephalopathy on magnetic resonance imaging: a case report. BMC Neurol 2021;21:156. 10.1186/s 12883-021-02182-z 33838643 PMC 8035775 · doi ↗ · pubmed ↗
- 6Mateen FJ, Keegan BM, Krecke K. et al. Sporadic leucodystrophy with neuroaxonal spheroids: persistence of DWI changes and neurocognitive profiles: a case study. J Neurol Neurosurg Psychiatry 2010;81:619–22. 10.1136/jnnp.2008.16924320176606 · doi ↗ · pubmed ↗
- 7Gavrilova RH, Raghunathan A, Flanagan EP. et al. Inflammatory leukoencephalopathy mimicking hereditary disease. Neuroimmunol Rep 2022;2:100092. 10.1016/j.nerep.2022.100092 · doi ↗
- 8Lynch DS, Zhang WJ, Lakshmanan R. et al. Analysis of mutations in AARS 2 in a series of CSF 1R-negative patients with adult-onset leukoencephalopathy with axonal spheroids and pigmented glia. JAMA Neurol 2016;73:1433–9. 10.1001/jamaneurol.2016.222927749956 · doi ↗ · pubmed ↗