Divergent Organomagnesium Reactivity of Rigid, Dinucleating Naphthyridine Ligands: Backbone Changes with Big Impact
Errikos Kounalis, Marieke M. Broekman, Puck Uyttewaal, Uladzislava Dabranskaya, Martin Lutz, Daniël L. J. Broere

TL;DR
This paper explores how small changes in ligand structure significantly affect the reactivity and stability of organomagnesium complexes.
Contribution
The study reveals how ligand architecture influences steric strain and reactivity in dimagnesium complexes.
Findings
Amide-based ligands reduce steric strain around the Mg2Cl2 core.
Secondary amine ligands form stable complexes, while amide ligands trigger radical reactions.
Ligand architecture profoundly impacts coordination chemistry and reactivity.
Abstract
We report the synthesis and characterization of two naphthyridine-based ligands bearing pendant secondary amine and amide donors, respectively. We additionally report their deprotonation chemistry and reactivity with dialkylmagnesium and Grignard reagents. The Grignard reactions yield structurally distinct LMg2Cl2·(THF) n complexes, with the amide-based complex exhibiting reduced steric strain from the ligand around the Mg2Cl2 core. Comparison of the steric profiles of the LMg2Cl2·(THF) n complexes reveals that this reduced steric strain stems from the difference in binding modes of the ligands, which in the amide case points the bulk of sterically demanding substituents away from the Mg2Cl2 core. Reactivity of the ligands with Mg(n-Bu)2 shows divergent outcomes: the secondary amine-based ligand forms the LMg2(n-Bu)2·(THF)2 complex cleanly, whereas the amide-based ligand produces…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
 1
1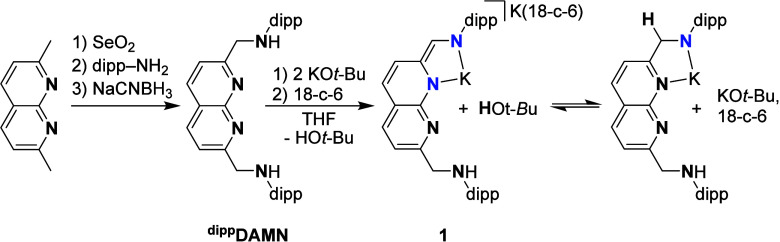 2
2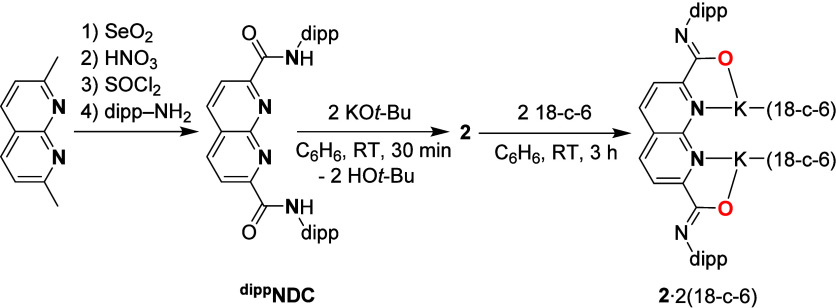 3
3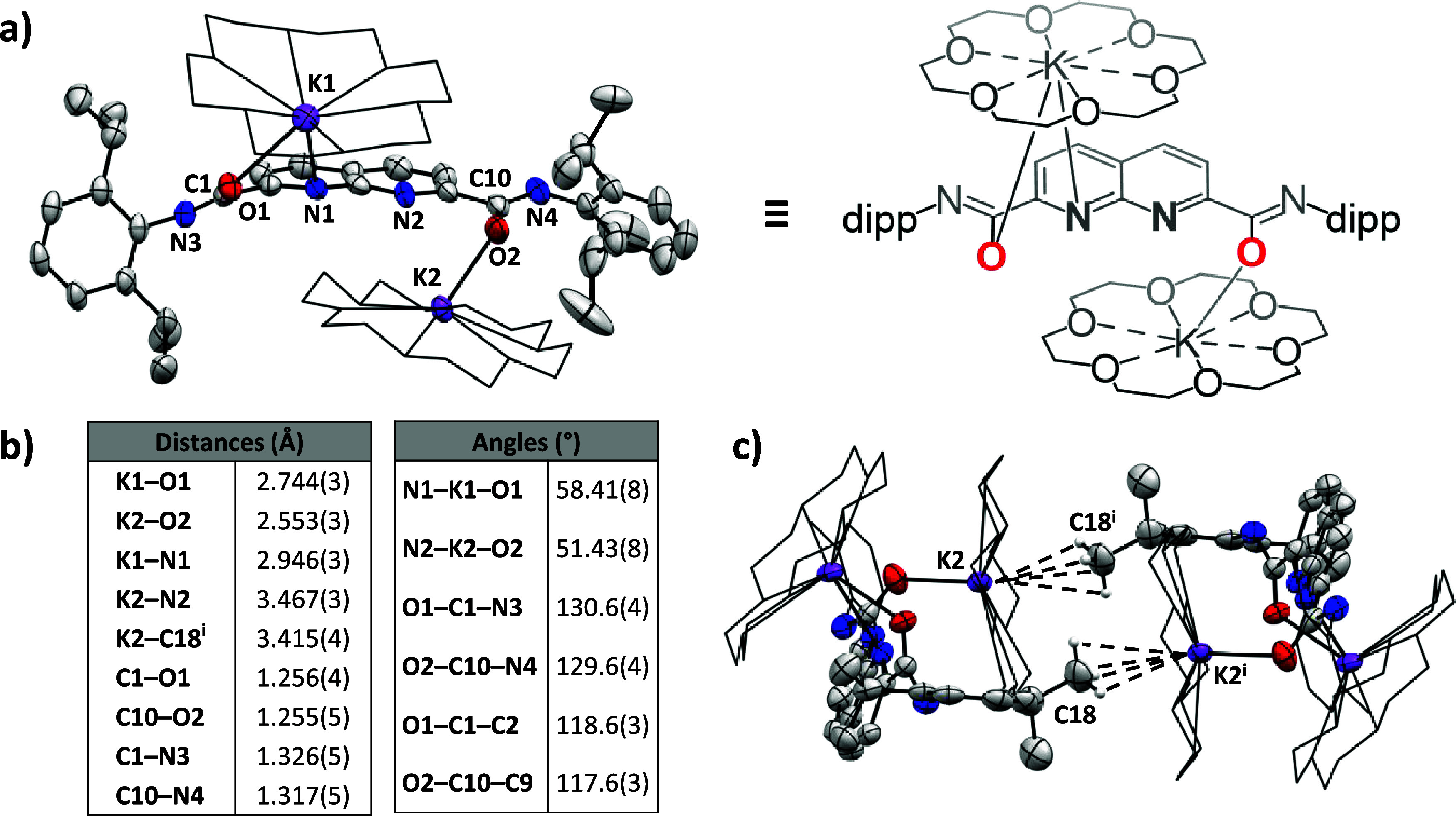 1
1 4
4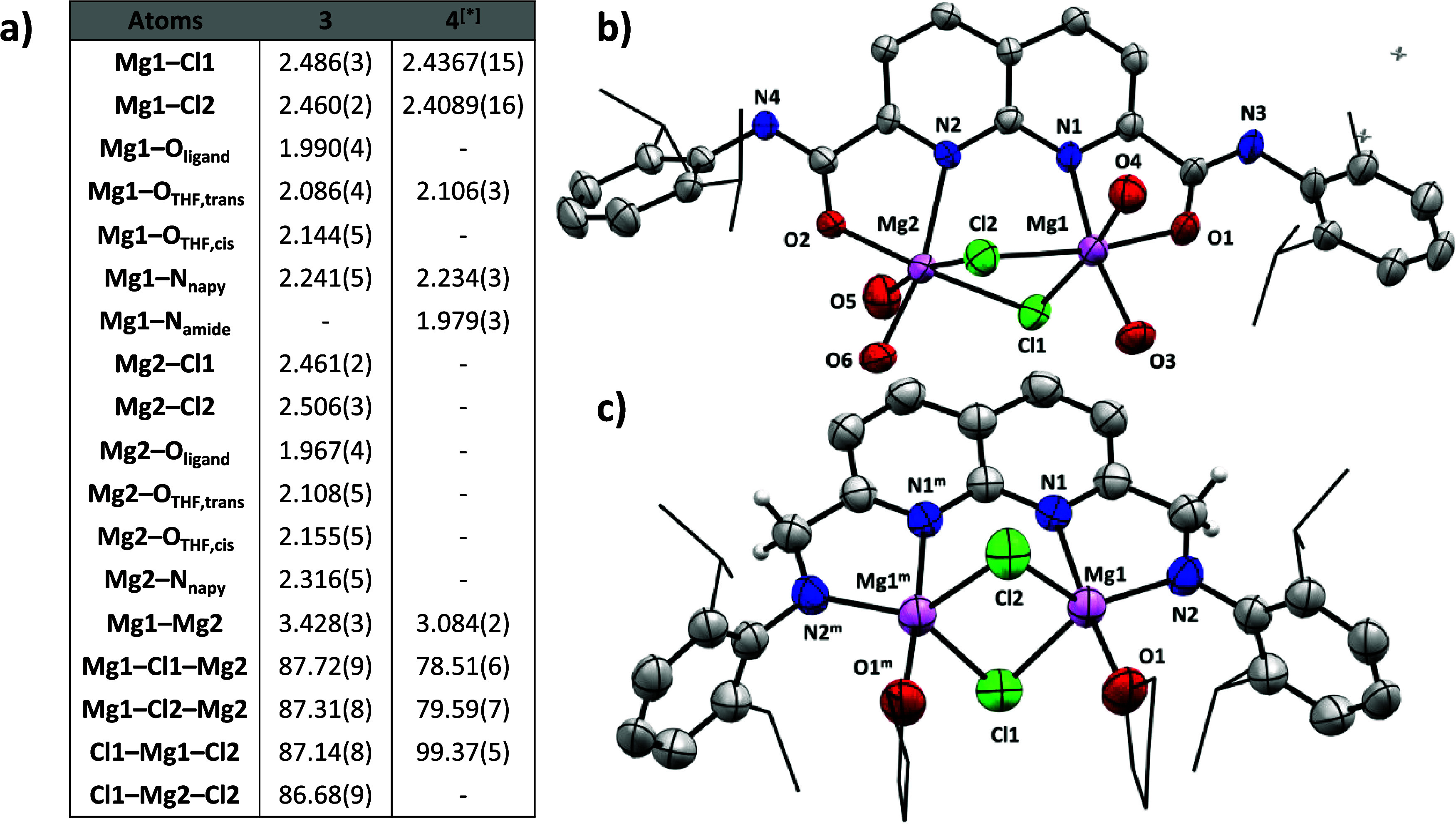 2
2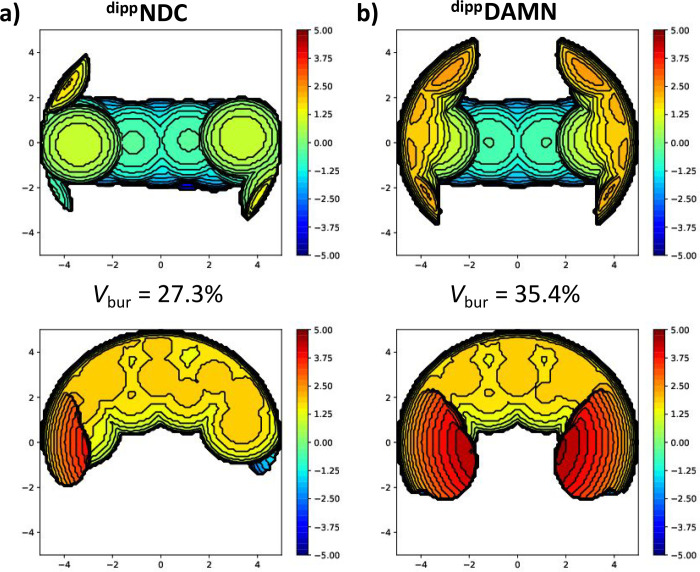 3
3 5
5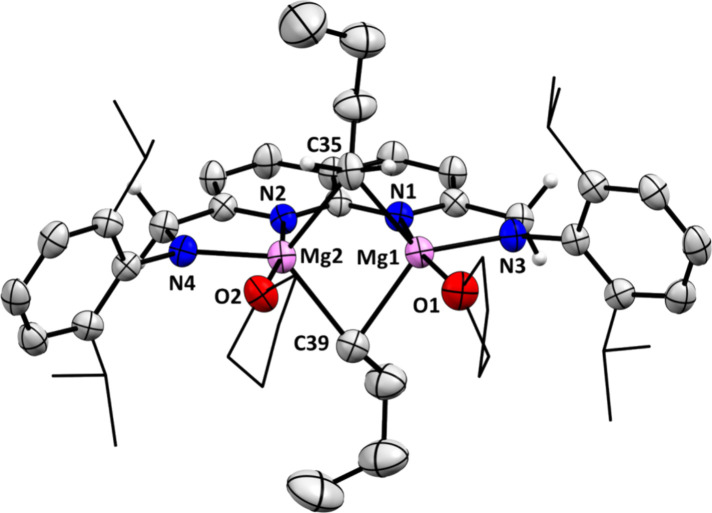 4
4 6
6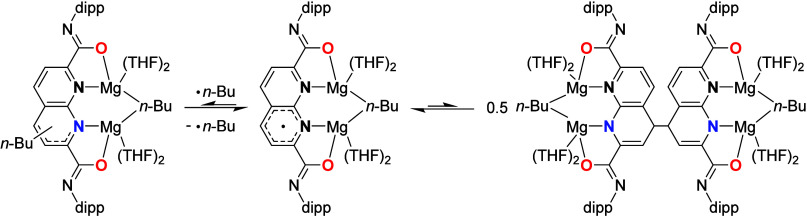 7
7- —Nederlandse Organisatie voor Wetenschappelijk Onderzoek10.13039/501100003246
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsCoordination Chemistry and Organometallics · Organometallic Complex Synthesis and Catalysis · Cyclopropane Reaction Mechanisms
Introduction
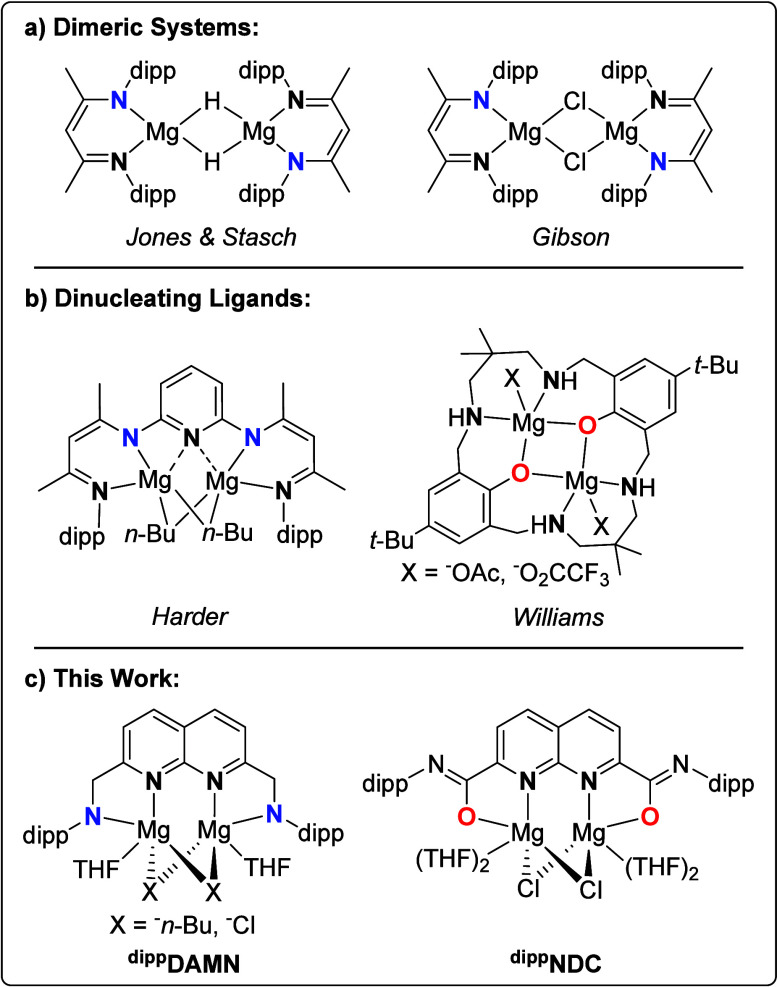
The high abundance of magnesium in the Earth’s crust, coupled with its low toxicity,? has driven considerable interest in exploring the potential of Mg-based complexes for various applications. Extending beyond the well-established organomagnesium chemistry pioneered by Grignard,? Mg complexes have demonstrated utility in diverse reactions, including hydroelementation, ?−? ? ? ? ? Lewis-acid catalysis, ?,? polymerization,? and dehydrocoupling.? These complexes are often stabilized by anionic diketiminate (NacNac) ligands, which employ hard nitrogen donors to support the Mg centers.? The propensity for dimerization (resulting in a Mg_2_X_2_ “diamond core”) in such systems is well-documented, with a variety of bridging ligands, such as halides, ?−? ? hydrides,? amidoboranes,? and alkyl groups, ?,? forming dimers that mitigate the electronic and coordinative unsaturation of monomeric species. The nuclearity of Mg complexes, influenced by factors such as steric strain, ?,? the presence of coordinating solvents or coligands, ?,?,?−? ? and coligand size,? significantly impacts their reactivity. For example, computational studies by the Maron and Hill groups on Mg-catalyzed hydrosilylation of alkenes have shown that dinuclear complexes can retain their nuclearity during alkene insertion, while subsequent steps involve monomeric species.? The cooperative reactivity of these dinuclear complexes has been highlighted in work by the Harder group, where dimagnesium complexes based on dinucleating (bis-NacNac) ligands were shown to result in an alternative mechanistic pathway and decreased onset-temperatures for the dehydrocoupling of amidoborane complexes.? Additionally, the Williams group has demonstrated that a dimagnesium complex bound to a macrocyclic ligand is highly active toward epoxide/CO_2_ ring-opening copolymerization (ROCOP).? Despite these advantages, the flexibility of the ligands in these examples often allows the Mg centers to adopt variable distances, complicating efforts to study the intrinsic reactivity of the dimagnesium core.
To address this challenge and harness the advantages of enforced nuclearity, we aimed to synthesize dimagnesium complexes within a rigid, dinucleating framework. We chose the 1,8-naphthyridine motif as the basis for our ligands, as this framework provides a constrained environment to enforce close proximity between the Mg centers and suppresses monomer–dimer equilibria that could alter reactivity.? Previous work has shown that the phosphine donors of the naphthyridine-based bis-phosphine ‘expanded pincer’ ligand (^ t ‑Bu ^ PNNP) developed by our group can be a poor match for hard metals.? As such, in this work, we introduce the Di-Amino-Methylene-Naphthyridine (^ dipp ^ DAMN) and Naphthyridine Di-Carboxylamide (^ dipp ^ NDC) ligands (Schemec). Both ligands feature anionic flanking donors, making them more suitable for stabilizing dinuclear cores of harder metals such as Mg. We report the deprotonation chemistry of both ligands and detail the synthesis and characterization of various dimagnesium complexes supported by these ligands and quantify their steric profiles. These studies expand the library of dimagnesium complexes and offer insights into how nuclearity and ligand design influence magnesium-based coordination chemistry and reactivity, displaying markedly divergent behavior upon modifications in the ligand architecture.
*(a) Representative Dimeric Dimagnesium Complexes Reported in Literature, ,
(b) Dimagnesium Complexes Supported by Dinucleating Ligands, , and (c) the Dimagnesium Complexes Reported in This Work*
Results and Discussion
Synthesis of dippDAMN and Its Deprotonation
The ^ dipp ^ DAMN ligand (Scheme) was synthesized as a slightly air-sensitive white solid in three steps from 2,7-dimethyl-1,8-naphthyridine in a 32% overall yield (see ESI Section 1.2). We envisioned that isolation of the dipotassium salt of the ligand would allow access to complexes of metals where internal base precursors are not available, similar as demonstrated for the related ^ t ‑Bu ^ PNNP ligand.? Accordingly, the ^ dipp ^ DAMN ligand was treated with two equiv of KOt-Bu in THF, which led to a dark orange solution. Analysis of the reaction mixture by ^1^H NMR (toluene-d 8, 298 K) spectroscopy revealed broad resonances, which we attributed to fluxional binding of K^+^. Using VT NMR spectroscopy, no decoalescence was observed down to 193 K (toluene-d 8, see ESI Figure S7). Notably, the addition of 1 equiv 18-crown-6 to this mixture resulted in a color change to dark green and the concomitant formation of 1. The ^1^H and ^13^C{^1^H}NMR spectra (C_6_D_6_, 298 K) showed a number of resonances indicative of the loss of the C_2v_-symmetry (see ESI Section 1.4). The ^1^H NMR spectrum revealed four equally integrating naphthyridine resonances (δ = 6.34–5.78 ppm) at a more upfield shift compared to the free ligand, which agrees with partial dearomatization of the naphthyridine core. This is further confirmed by the observation of a singlet at δ = 5.46 ppm which we attribute to the methine proton. The presence of a broader apparent singlet at δ = 4.84 ppm (attributed to N–H) and a doublet (^3^ J H,H = 4.8 Hz) at δ = 3.98 ppm (attributed to –CH 2) integrating in a 1:2 ratio is consistent with one side of the naphthyridine motif being doubly deprotonated and the other side being left intact. One N–H proton being still present in 1 could be additionally confirmed through the observation of a weak intensity vibration at 3502 cm^–1^ in the ATR-IR spectrum. Deprotonation of the free ligand in the related ^ t ‑Bu ^ PNNP ligand can result in either the cis-isomer (i.e., the donor arm pointing toward the binding pocket) or the trans-isomer.? To gauge which of the isomers was obtained in this case, a 2D-NOESY spectrum was recorded of 1. The antiphase cross-peak between the methine resonance and one the naphthyridine resonances at δ = 6.02 ppm confirmed the formation of the cis-isomer (see ESI Figure S10). The same 2D-NOESY however also showed an in-phase cross-peak (EXSY) between the methine resonance and a very broad resonance at δ = 6.63 ppm. Recording VT ^1^H NMR spectra of a toluene-d 8 solution of 1 at lower temperatures revealed sharpening of this resonance with a clear peak being visible at temperatures below 233 K (see ESI Figure S14). This allowed for integration, showing a 1:1 ratio with the methine resonance.
*Synthesis of the Dinucleating dipp
DAMN and Subsequent Deprotonation, Resulting in Complex 1 and the Observed Equilibrium with the Protonated Form*
We attribute this broad resonance to the O–H proton of a t-BuOH molecule which reversibly protonates the methine carbon of 1, resulting in the equilibrium depicted in Scheme. A closer inspection of the ^1^H NMR spectrum revealed a singlet at δ = 1.35 ppm, partially obfuscated by the dipp methyl resonances, which could potentially be attributed to the t-Bu protons of the exchanging t-BuOH. Extensive (freeze-)drying of the compound did not lead to a decrease of the amount of exchanging t-BuOH, suggesting that in the solid state the equilibrium lies to the side of the nondearomatized ligand (Scheme, right). Interestingly, addition of an extra equiv of 18-c-6 did not result in the change of speciation, suggesting that one K^+^ ion is tightly bound in the naphthyridine pocket as depicted in Scheme.
Combined, these observations show that upon deprotonation of the secondary amine, the methylene linker on the same side becomes more acidic and gets subsequently deprotonated by the second equiv of KOt-Bu. This unexpected deprotonation of the ^ dipp ^ DAMN ligand led us to set our sights on a ligand design without methylene protons. By substituting the pendant secondary amines with pendant amides, we expected to retain the binding features ^ dipp ^ DAMN while suppressing the observed deprotonation of the methylene linkers.
Ligand Synthesis and Deprotonation Studies of dippNDC
Consequently, the ^ dipp ^ NDC ligand (Scheme) was synthesized in 4 steps from 2,7-dimethyl-1,8-naphthyridine in an overall 44% yield (see ESI Section 1.5).? ^ dipp ^ NDC was treated with two equiv of either KOt-Bu or KBn in C_6_H_6_ (Scheme, middle). Analysis of the reaction mixture (out of KBn) in C_6_H_6_ by ^1^H NMR spectroscopy (298 K, PRESAT) before removal of the solvent revealed clean conversion to a new species (2, see ESI Figure S28). The number of resonances observed are consistent with a species symmetric on each side of the mirror plane perpendicular to the naphthyridine plane. A singlet resonance attributed to the amide protons is absent, suggesting full deprotonation of the ligand. For the –CH 3 protons on the dipp substituents, two doublets at δ = 1.32 and 1.20 ppm integrating to 12 H each were observed and for the methine protons only one resonance integrating to 4 H was observed. This difference in magnetic environment of the –CH 3 groups can be indicative of slow or hindered rotation of the N–C_Ar_ bonds on the NMR time scale, rendering these the –CH 3 groups diastereotopic. While removal of the solvent of 2 resulted in the irreversible formation of an intractable mixture of products, the addition of 2 equiv of 18-crown-6 enabled the isolation of 2·2(18-c-6) (Scheme). Inspection of the ^1^H NMR spectrum (in C_6_D_6_ at 298 K, see ESI Figure S29) shows a shift of the resonances compared to 2, indicative of a change in the binding of K^+^. Additionally, the appearance of the – CH 3 protons as a single (albeit broad) doublet at δ = 1.60 ppm which integrates to 24 H, suggests that the previously proposed constrained rotation of the N–C_Ar_ bonds might be alleviated upon sequestration of K^+^. Layering a saturated toluene solution of 2·2(18-c-6) with pentane resulted in the formation of single crystals, suitable for X-ray diffraction. Elucidation of the crystal structure (Figure) revealed the dimeric nature of 2·2(18-c-6) in the solid-state. Two independent crown ether-sequestered K^+^ ions are found per naphthyridine motif, residing above and below the naphthyridine plane, respectively. The ^ dipp ^ NDC ligands are bound to K^+^ via the imidate (via oxygen) binding mode rather than the amidate (via nitrogen) binding mode,? with K–O_(lig)_ distances of 2.553(3) Å and 2.744(3) Å. There is a short contact between one of the sequestered potassium cations and a methyl group on one of the dipp groups of the other molecule in the dimer. This short K···C contact of 3.415(4) Å is within the range of reported examples of – CH_3_ to K^+^ agostic bonding where the K^+^ is sequestered by 18-crown-6. ?−? ? ?
*Syntheses of the dipp
NDC Ligand, and Complexes 2 and 2·2(18-c-6)*
a) Displacement ellipsoid plot of 2·2(18-c-6) at 50% probability, flanked by a schematic representation of the perspective. Hydrogen atoms, minor disorder components and cocrystallized toluene are omitted for clarity. Additionally, only half of the dimer is depicted, and 18-crown-6 molecules are depicted as wireframe to enhance clarity. b) Selected bond distances and angles in Å and °, respectively. Symmetry code i: – x, 1–y, 1–z. c) Side-view of the dimer of 2·2(18-c-6), visualizing the agostic interactions between K+ and one of the –CH3 substituents on one of the dipp groups.
dippNDCMg2Cl2
We hypothesized that transmetalation of 2·2(18-c-6) with metal precursors would most likely result in the retention of the imidate binding mode (see below). To investigate if we could access the amidate form of the ligand, we attempted direct metalation of the N–H bond using Grignard reagents, which contain an internal base. A reaction between ^ dipp ^ NDC and two equiv MeMgCl resulted in a bright orange solution concomitant with visible effervescence. From this mixture, 3 was isolated as a yellow solid in 74% yield (Scheme, left). Analysis of the ^1^H and ^13^C{^1^H} NMR spectra (THF-d 8, 298 K) revealed full conversion of the ligand to a new compound with a number of resonances consistent with a C_2v_-symmetry on the NMR time scale (see ESI Section 1.7). The observed symmetry and absence of N–H resonances are in line with full deprotonation of the ^ dipp ^ NDC ligand. Layering a saturated solution of 3 in THF with hexane and storing it at –40 °C for several days yielded orange single crystals suitable for analysis by X-ray diffraction.
Syntheses of Complexes 3 and 4
The solid-state structure of 3 (^ dipp ^ NDCMg_2_Cl_2_·4THF, Figureb) revealed the formation of a dinuclear complex containing two independent Mg centers, one in each of the binding pockets of the ^ dipp ^ NDC ligand. Similar to 2·2(18-c-6), the imidate binding mode was obtained in the solid-state structure rather than the amidate binding mode, suggesting that the ligand readily tautomerizes upon deprotonation. Two THF molecules (cis to each other) are bound to each Mg center and together with the bridged chloride ligands a distorted octahedral geometry around Mg is obtained. The two Mg centers are positioned slightly outside of the naphthyridine plane (see ESI Figure S62), with the orientation of the axial ligands per center being antiparallel. The combination of this geometry with the variety of ligands on the Mg centers results in optical isomerism, with both the Λ,Λ and Δ,Δ compounds being formed in solution. The analyzed crystal was comprised of a single enantiomer of 3 (Δ,Δ). Having obtained structural insights and spectroscopic handles of 3, we reacted 2·2(18-c-6) with 2 equiv of MgCl_2_ in THF. ^1^H NMR analysis of the reaction mixture showed 3 as a major product, thereby confirming our aforementioned hypothesis that this reaction would yield the imidate tautomer (see ESI Figure S36).
a) Selected bond distances and angles of compounds 3 and 4 in Å and °, respectively. [∗] 4 is located on a mirror plane. Only half of the distances and angles are independent. b) Displacement ellipsoid plot of the Δ,Δ enantiomer of 3 at 50% probability. Hydrogen atoms, the carbon backbones of coordinated THF and minor disorder components are omitted for clarity. The isopropyl groups on the dipp substituents are depicted as wireframe for clarity. c) Displacement ellipsoid plot of 4 at 50% probability. Most hydrogen atoms, severely disordered solvent molecules, and the minor disorder components of THF are omitted for clarity. The isopropyl groups on the dipp substituents are depicted as wireframe for clarity. Symmetry code m: x, 1/2–y, z.
dipp
DAMNMg2Cl2·2 THF
The imidate binding mode found for 2 and 3 decreases the steric pressure around the dimagnesium core due to steric bulk of the dipp groups pointing away from the dinuclear core. In order to investigate the influence the decrease in steric bulk around the Mg_2_Cl_2_ core has on its coordination chemistry, we targeted the synthesis of the Mg_2_Cl_2_ complex of the ^ dipp ^ DAMN ligand for direct comparison with 3. Addition of ^ dipp ^ DAMN to a THF solution of 2 equiv of MeMgCl led to clean formation of 4 as a bright orange solid in quantitative yield (Scheme, right). Analysis of the ^1^H NMR and ^13^C{^1^H} NMR spectra (C_6_D_6_, 298 K) showed a number of resonances consistent with retention of the C_2v_-symmetry of the ligand (see ESI Section 1.9). The loss of the N–H resonances, the presence of only two naphthyridine resonances (δ = 7.00 and 6.61 ppm) and the appearance of a singlet at δ = 5.08 ppm integrating to 4H, which we attribute to the methylene linkers, all suggested that the secondary amines are selectively deprotonated by the methyl groups of the Grignard reagent, contrasting the deprotonation order observed for 1. In addition, broad resonances are observed at δ = 3.65 and 1.25 ppm suggesting that THF is bound to the Mg centers in solution.
Layering a saturated THF solution of 4 with pentane at –40 °C led to the formation of orange needles suitable for analysis by X-ray diffraction. The solid-state structure of 4 (Figurec) agrees with the spectroscopic data and confirms the selective deprotonation of the N–H protons by the Grignard reagent, with each Mg center residing in a bidentate binding pocket of the dinucleating ligand. In the crystal, 4 is located on a mirror plane. The two chlorides bridge the two Mg centers in a “diamond core” fashion and a THF ligand per Mg completes the observed trigonal bipyramidal geometry (τ = 0.75), with the naphthyridine N-donor and the THF O-donor occupying the axial positions. The bond between the naphthyridine N-donor and Mg (Mg1–N1 = 2.234(3) Å) is substantially longer than the bond between Mg and the pendant amide (Mg1–N2 = 1.979(3) Å), consistent with the neutral and anionic character of these donors, respectively.?
Structural Insights
With the solid-state structures of the LMg_2_Cl_2_ complexes determined, a comparison of the structural features and the bond metrics is possible. The most striking difference between the two complexes is the geometry around the Mg centers (Figure), being octahedral in 3 and trigonal bipyramidal in 4. We ascribe this change in geometry to the imidate binding mode of the ^ dipp ^ NDC ligand in 3, which results in a decrease of steric bulk around the Mg centers (for a quantification of the steric bulk in both complexes see below). The decreased steric encumbrance allows for the binding of an additional THF molecule and implies that the Mg centers in 4 are coordinatively unsaturated. The increased steric and electronic saturation in 3 results in longer Mg–Cl bonds compared to the same bonds found for 4 (2.460(2)-2.506(3) Å in 3 vs 2.4089(16)-2.4367(15) Å in 4). The binding of an additional THF molecule in 3 also has a significant influence on the Mg···Mg distance. This distance in 3 is significantly larger than the distance in 4 (3.428(3) Å vs. 3.084(2) Å respectively). This is due to one of the Mg centers in 3 being pushed outside of the naphthyridine plane and one slightly below the naphthyridine plane (as is evident from their respective torsion angles χ[O1,C1,C2,N1]= – 10.6(8)° and χ[O2,C10,C9,N2]= – 2.7(8)°, see ESI Figure S62) to minimize the distortion of the octahedral geometry. The distance of Mg1 to the least-squares plane of the naphthyridine ring is – 0.897(2) Å and the corresponding distance for Mg2 is 0.495(2) Å. Contrastingly, the Mg centers in 4 reside in the naphthyridine plane, with the distance of Mg1 to the least-squares plane of the naphthyridine ring being 0.0799(10) Å. To quantify the steric environments surrounding the Mg_2_ cores in 3 and 4 imposed by each of the ligands, we used the SambVca 2.1 web application to build their respective steric maps.? For dinuclear systems, choosing the centroid and radius of the defined sphere for the buried volume calculations is nontrivial, and based on previous work from our group we placed the centroid of the sphere in between the metal centers and set the radius of the defined sphere to 5 Å.? The steric maps in Figure reveal that the largest difference in steric encumbrance in the reaction hemisphere of the two complexes is in the cis ligand void space (i.e., above and below the naphthyridine plane).? In the case of 4, the i-Pr moieties on the dipp substituents shield the Mg centers above and below the naphthyridine plane, preventing the coordination of additional THF molecules. In the case of 3, the dipp substituents are oriented away from the Mg centers, which significantly decreases the shielding of the cis void spaces by the i-Pr groups. The difference in the steric properties of the ligands in 3 and 4 are also expressed in their respective buried volumes (V bur) of 27.3% and 35.4%. We envision that these pronounced differences in steric profiles between the two complexes can lead to diverging reactivity due to the varying accessibility of the Mg centers for substrates.
*Steric maps and associated buried volumes of a) the dipp
NDC ligand as bound to 3 and b) the dipp
DAMN ligand as bound to 4. The top maps show the front view of the ligand systems (parallel with the naphthyridine plane), and the bottom maps show the top-down view of the ligand systems (perpendicular to the naphthyridine plane).*
dipp
DAMNMg2(n-Bu)2·2 THF
The discrepancy in deprotonation order observed for 1 and 4, along with the relative acidity of the methylene protons, prompted us to investigate whether treating ^ dipp ^ DAMN with two equiv of a dialkyl-magnesium species would result in deprotonation and subsequent dearomatization of the ligand, similar to what is observed for the related ^ t ‑Bu ^ PNNP ligand when treated with strong bases. ?,? Treating a THF solution of ^ dipp ^ DAMN with 2 equiv of Mg(n-Bu)2 (Scheme) resulted in clean formation of 5 as a brown solid in quantitative yield. The number of resonances in the ^1^H- and ^13^C{^1^H}-NMR spectra (see ESI Section 1.10) is consistent with a C_2v_-symmetric compound. Broad resonances at δ = 3.37 and 1.18 ppm (C_6_D_6_, 298 K), integrating to 8H, are indicative of 5 binding two THF molecules in solution. Additional resonances at δ = 1.49 (overlapping with the –CH(CH 3)2 resonance of the dipp group, 4H), 1.31 (4H), 0.90 (6H) and the characteristic AA’BB’ resonance at 0.01 ppm (4H) are consistent with two butyl groups being present in the complex.
*Reactions of Mg(n-Bu)2 with the dipp
DAMN and dipp
NDC Ligands, Resulting in the Formation of 5 and a NMR-Silent Species, Respectively*
Storing a saturated THF/pentane solution of 5 at – 40 °C yielded dark needle-shaped crystals suitable for X-ray diffraction (Figure). The solid-state structure revealed a distorted trigonal bipyramidal geometry around the Mg centers, with each butyl group bridging the Mg centers. The apical positions of the bipyramids are occupied by the N-donor of the naphthyridine ring and by a O-donor from THF. The 1:1 ratio between coordinated THF and Mg in the solid-state structure is consistent with earlier observations in solution. The N3–Mg1–C35 and N3–Mg1–C39 angles (125.10(15)° and 129.42(15)° respectively) are enlarged, while the C35–Mg1–C39 angle (104.92(15)°) is contracted compared to ideal trigonal bipyramidal geometry, with similar trends observed for the second Mg center (τ = 0.65 and 0.71, respectively). The Mg1–Mg2 distance (2.7499(18) Å) and Mg–C bond lengths (2.302(4)-2.321(4) Å) align with metrics reported for crystallographically characterized Mg_2_–alkyl_2_ species with a similar “diamond-core”. ?,?,?,?−? ? ? ? ? The observation of the preferential formation of this core over deprotonation of the relatively acidic methylene protons is most likely attributed to the bridging binding mode of the butyl groups, which delocalizes their negative charge and renders them less basic. The Mg–C bond lengths are slightly longer than in most of these structures and likely reflect the THF coordination and higher electronic saturation of the Mg centers. The bond lengths of Mg1–N3 and Mg2–N4 (2.022(3) and 2.020(4) Å respectively) are considerably shorter than the Mg1–N1 and Mg2–N2 bonds (2.247(3) and 2.220(3) Å respectively), which is in line with their higher anionic character.
Displacement ellipsoid plot of 5 at 50% probability. Most hydrogen atoms, cocrystallized solvent molecules and minor disorder components are omitted and parts of the THF molecules and the dipp substituents on N are depicted as wireframe for clarity. The hydrogen atoms on C35 were found in the difference Fourier maps.
dipp
NDCMg2(n-Bu)2
We explored if the dibutyl-magnesium chemistry observed for the ^ dipp ^ DAMN ligand also translated to the ^ dipp ^ NDC ligand. Reacting a benzene solution of ^ dipp ^ NDC with 2 equiv of Mg(n-Bu)2 resulted in effervescence and a dark green solution. Interestingly, ^1^H NMR (PRESAT) analysis of the reaction mixture revealed the formation of an NMR-silent species (see ESI Figure S49), contrasting the described reactivity with ^ dipp ^ DAMN. X-band EPR spectroscopic analysis of the reaction mixture revealed an intense isotropic signal (g iso = 2.0036) without well-resolved hyperfine interactions, consistent with the presence of paramagnetic species (Figure S50–S52). Despite repeated attempts, we were unable to grow single crystals suitable for analysis by X-ray diffraction out of the reaction mixture. To better understand the nature of the complexes formed, we quenched the reaction mixture with water (see ESI Section 1.11). ^1^H NMR analysis (C_6_D_6_, 298 K) after the aqueous workup revealed multiple resonances, consistent with the formation of several compounds. Of particular interest were the resonances between δ = 5.91 and 4.85 ppm, consistent with protons on dearomatized pyridine rings. ?,? The ^1^H–^1^H COSY NMR spectrum (see ESI Figure S55) showed coupling between the resonance at δ = 4.85 ppm and a doublet at δ = 3.23 ppm, obfuscated by the -dipp – CH septets. These shifts are similar to those reported for the 3- and 4-positions of a para-coupled bis-naphthyridine system (δ = 5.0 and 3.4 ppm respectively, in toluene-d 8).? Such para-coupling arises from the radical recombination of naphthyridine-centered radicals, as observed for dinickel complexes (Uyeda group) and dicopper complexes (Tilley group). ?,? Homonuclear and heteronuclear 2D NMR spectra (see ESI Figures S57–58) reveal that these resonances attributed to a para-coupled bis-naphthyridine arise from a distinct molecule. The remaining resonances in the δ = 5.91–4.85 ppm region correspond to a molecule bearing a butyl group, as evidenced by characteristic resonances at δ = 1.96 (α–CH 2) and 0.82 ppm (−CH 3). ESI-MS analysis (positive mode, see ESI Figure S59) of the reaction mixture postaqueous workup revealed signals at m/z = 592.5, 617.2 and 1195.8, which can be attributed to the [M]^+^ (M = ^ dipp ^ NDC ^ Bu ^), [M + Na]^+^ (M = ^ dipp ^ NDC ^ Bu ^ H), and [M+Na+MeCN]^+^ (M = ^ dipp ^ NDC ^ Bu ^ H– ^ dipp ^ NDCH), respectively (Scheme).
Some of the Identified Products That Form upon Quenching the NMR-Silent Reaction Mixture
Combining these data, we propose that a ^ dipp ^ NDCMg_2_(n-Bu)2 species initially forms, with the observed effervescence most likely arising from butane liberation. Consistent with previous observations, we expect that the ligand will be in the imidate form. This complex most likely undergoes Mg–C homolysis, similar to what is observed for dialkylmagnesium complexes bound to conjugated diimines, bis-pyridines, and di-iminopyridines.? DFT calculations on truncated LMg_2_Me_2_ models (see ESI Sections 2.4–2.7) reveal a Gibbs free energy difference of 38.3 kcal·mol^–1^ between the singlet and triplet states of the ^ dipp ^ DAMN-based complex. In contrast, the same difference for the ^ dipp ^ NDC-based complex is only 20.8 kcal·mol^–1^, making it thermally accessible at room temperature. Notably, the geometry optimization of the triplet state of ^ dipp ^ NDCMg_2_Me_2_·4 THF consistently resulted in extrusion of a methyl radical (see ESI Figure S61), highlighting the energetically facile nature of Mg–C homolysis in this case. For reported mononuclear complexes with pyridine-based ligands, homolysis of M–C bonds (M = Li, Mg, Al, Zn) often leads to alkylation of various positions of the pyridine, including the pyridinic nitrogen. ?,?,? This process is proposed to occur via single-electron transfer from the Mg–C bond to the pyridine backbone, followed by radical recombination with the liberated alkyl radical (Scheme, left). ?,? If the alkyl radical escapes the solvent cage, the resulting ligand-centered radicals can reversibly dimerize (Scheme, right). ?,? In these literature examples, both alkylation and dimerization products are diamagnetic, which contrasts with our observations for the ^ dipp ^ NDC ligand. We propose that the observed paramagnetism most likely results from the extended conjugated system of the ^ dipp ^ NDC ligand, which can stabilize ligand-centered radicals better than the mononuclear analogues, and pushes the alkylation and dimerization equilibria toward the ligand-centered radical side (Scheme, for a discussion of the proposed equilibria, see ESI Section 1.12). ?,?,? While the exact nature of the species formed in this reaction remains elusive, the combined NMR, EPR and ESI-MS data suggest a complex mixture of products, including butylated and coupled derivatives of the ^ dipp ^ NDC ligand. These products arise from the generated butyl radical being either trapped by or escaping the solvent cage, respectively.
Proposed Equilibria That Potentially Can Take Place upon Mg–C Homolysis
Conclusions
In conclusion, this work highlights the significant influence ligand architecture can have on the coordination chemistry of dimagnesium complexes supported by dinucleating naphthyridine-based ligands. For the described reactivity of the ^ dipp ^ NDC and ^ dipp ^ DAMN ligands with Grignard reagents this influence is of steric nature, with the steric bulk surrounding the Mg_2_Cl_2_ core differing markedly between the two ligands. This divergence is expressed in different coordination geometries around the Mg centers, which largely depend on the degree of coordinative saturation that is allowed by the steric bulk imposed by the dinucleating ligands. In contrast, the described reactivity of the ^ dipp ^ NDC and ^ dipp ^ DAMN ligands with dialkylmagnesium reagents highlights how changes in the ligand backbone can have a pronounced influence on the stability of the resulting Mg_2_Bu_2_ complexes. Whereas ^ dipp ^ DAMN forms a well-defined, diamagnetic Mg_2_(n-Bu)2 complex, the extended conjugated system of ^ dipp ^ NDC results in paramagnetic species that form upon Mg–C homolysis and subsequent radical reactivity. Collectively, these findings establish a framework for leveraging ligand design to tailor the reactivity of organomagnesium complexes, opening avenues for the development of novel organometallic transformations.
Supplementary Material
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Yaroshevsky A. A.Abundances of Chemical Elements in the Earth’s Crust Geochem. Int.2006441485510.1134/S 001670290601006 X · doi ↗
- 2Grignard V.Sur Quelques Nouvelles Combinaisons Organometalliques Du Magnesium et Leur Application a Des Syntheses d’alcools et d’hydrocarbures C. R. Acad. Sci.190013013221324
- 3Bauer, H. ; Harder, S. Early Main Group Metal Catalyzed Hydrogenation. In Early Main Group Metal Catalysis; John Wiley & Sons, Ltd, 2020; pp 175–199. 10.1002/9783527818020.ch 7. · doi ↗
- 4Bestgen, S. ; Roesky, P. W. Intramolecular Hydro amination of Alkenes. In Early Main Group Metal Catalysis; John Wiley & Sons, Ltd, 2020; pp 59–91. 10.1002/9783527818020.ch 3. · doi ↗
- 5Harder, S. Early Main Group Metal-Catalyzed Hydrosilylation of Unsaturated Bonds. In Early Main Group Metal Catalysis; John Wiley & Sons, Ltd, 2020; pp 151–173. 10.1002/9783527818020.ch 6. · doi ↗
- 6Sadow, A. D. Alkali and Alkaline Earth Element-Catalyzed Hydroboration Reactions. In Early Main Group Metal Catalysis; John Wiley & Sons, Ltd, 2020; pp 201–224. 10.1002/9783527818020.ch 8. · doi ↗
- 7Sarazin, Y. ; Carpentier, J.-F. Molecular S-Block Catalysts for Alkene Hydrophosphination and Related Reactions. In Early Main Group Metal Catalysis; John Wiley & Sons, Ltd, 2020; pp 93–121. 10.1002/9783527818020.ch 4. · doi ↗
- 8Magre M.Szewczyk M.Rueping M.Magnesium Complexes in Hydroelementation and Reduction Catalysis: Opportunities and Challenges Curr. Opin. Green Sustain. Chem.20213210052610.1016/j.cogsc.2021.100526 · doi ↗