Functional Characterization of KNOX and BELL Genes in Temperature-Responsive Floral Morphogenesis of Passion Fruit (Passiflora edulis)
Xinni Jiang, Jie Miao, Weifan Zu, Ruohan Zhou, Lexin Zheng, Ying Wei, Chunmei Lai, Rongjuan Qin, Ping Zheng, Xiuqing Wei, Jiahui Xu, Yuan Qin, Xiaoping Niu

TL;DR
This study explores how specific genes in passion fruit influence flower development under different temperatures, aiming to improve climate resilience in crops.
Contribution
The first genome-wide analysis of TALE transcription factors in passion fruit, revealing their roles in temperature-responsive floral development.
Findings
PeTALE21 regulates corona initiation, while PeTALE17 is active in later floral stages.
Cold stress upregulates PeTALE15/16/19/22 and heat suppresses PeTALE10/18/21.
PeTALE3/16/18/22/23 interact in a network that supports floral thermoresilience.
Abstract
Passion fruit (Passiflora edulis), a tropical crop of significant economic value, exhibits temperature-sensitive floral development. Here, we identified 23 TALE transcription factors (PeTALEs) and characterized their roles in floral organogenesis and thermal adaptation. Phylogenetic analysis classified PeTALEs into KNOX and BELL subfamilies, with conserved domain architectures and cis-regulatory elements linked to stress and hormone signaling. Spatiotemporal expression profiling revealed PeTALE21 as a key regulator of corona initiation, while PeTALE17 dominated in later floral stages. Temperature stress assays demonstrated cold-induced upregulation of PeTALE15/16/19/22 and heat-mediated suppression of PeTALE10/18/21. Yeast two-hybrid assays uncovered functional interactions between PeTALE3/16/18/22/23, highlighting a network governing floral thermoresilience. This study provides the…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
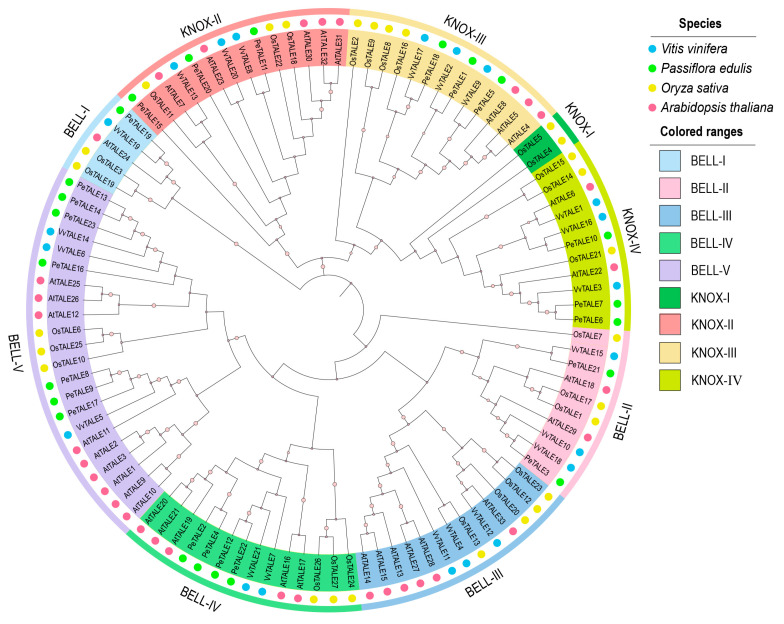 Figure 1
Figure 1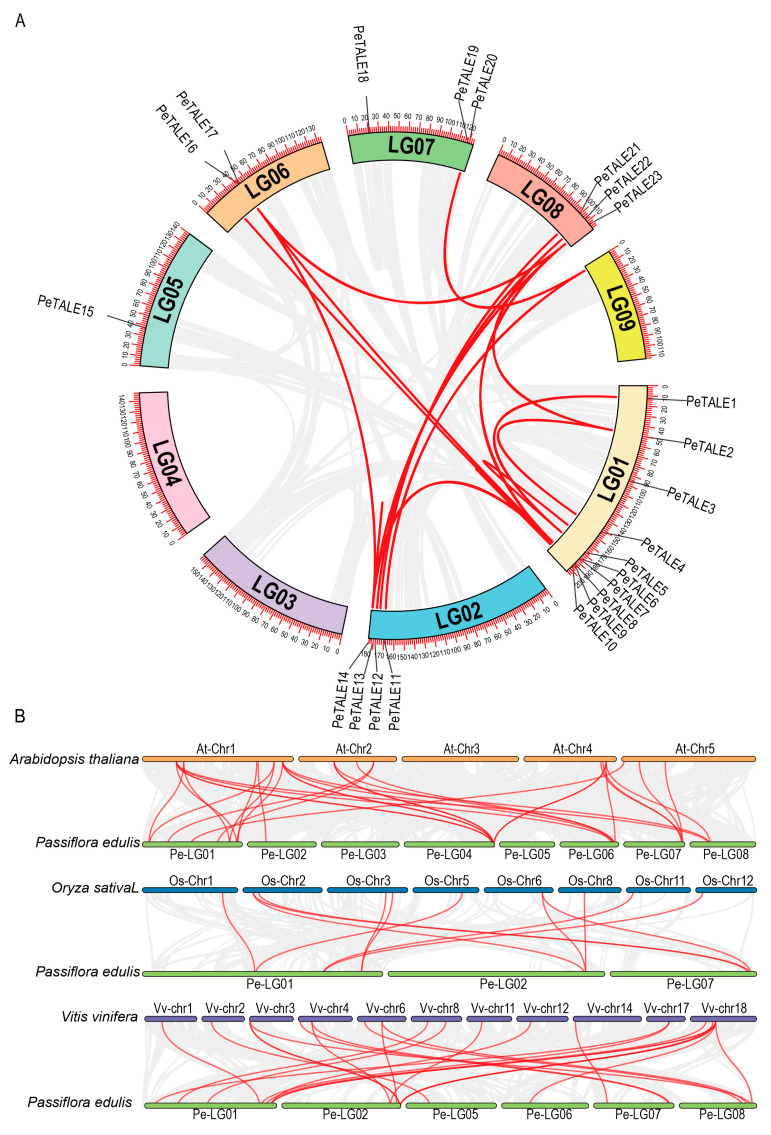 Figure 2
Figure 2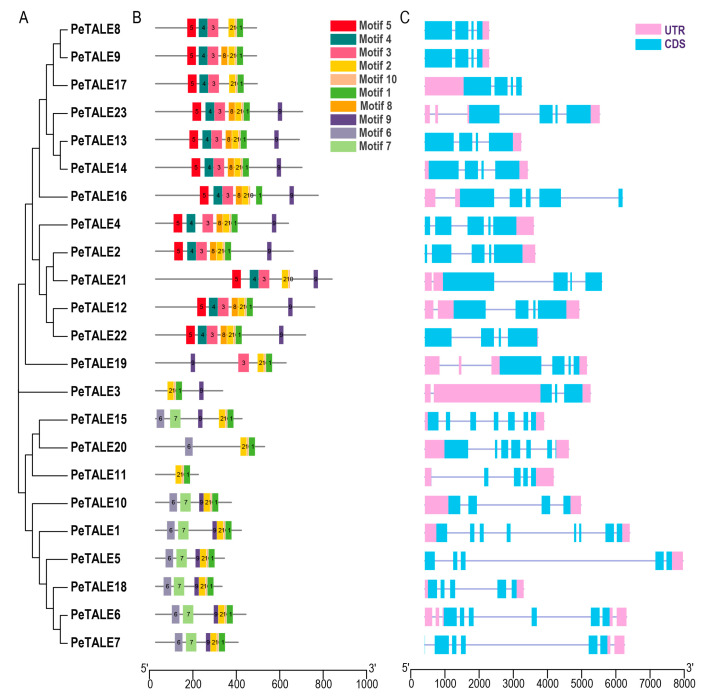 Figure 3
Figure 3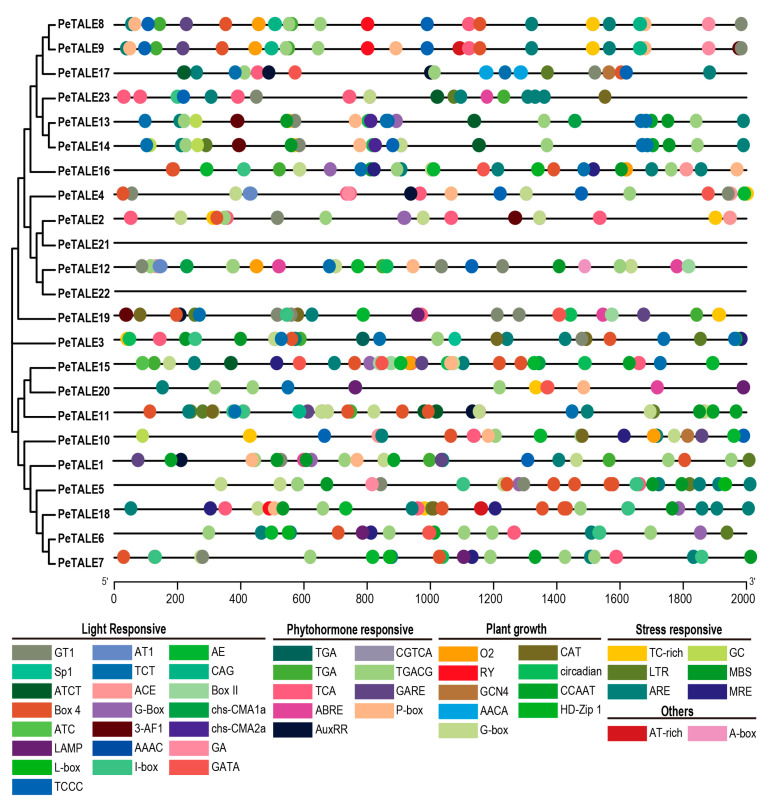 Figure 4
Figure 4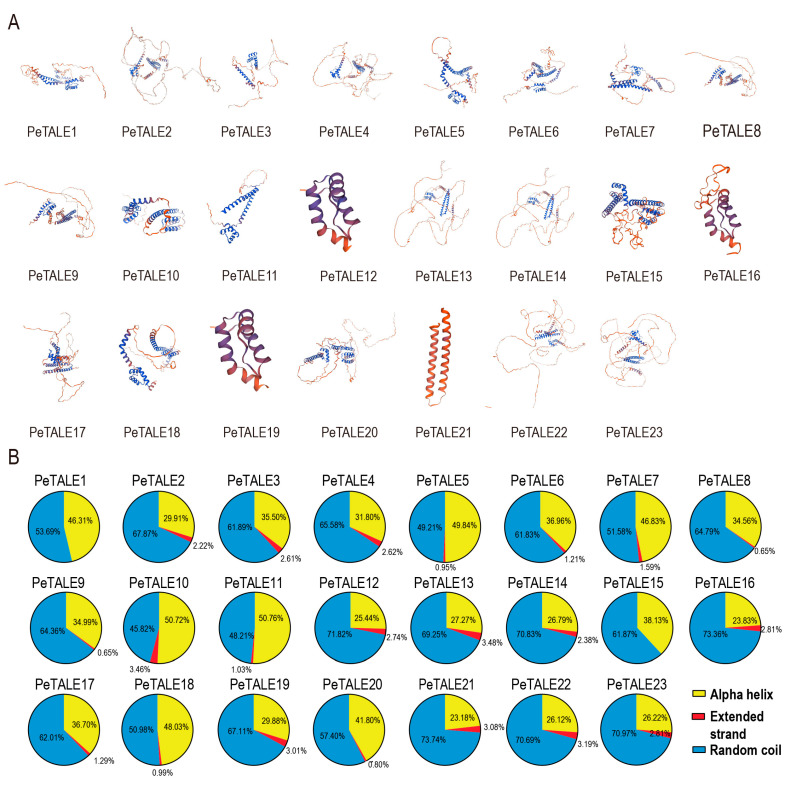 Figure 5
Figure 5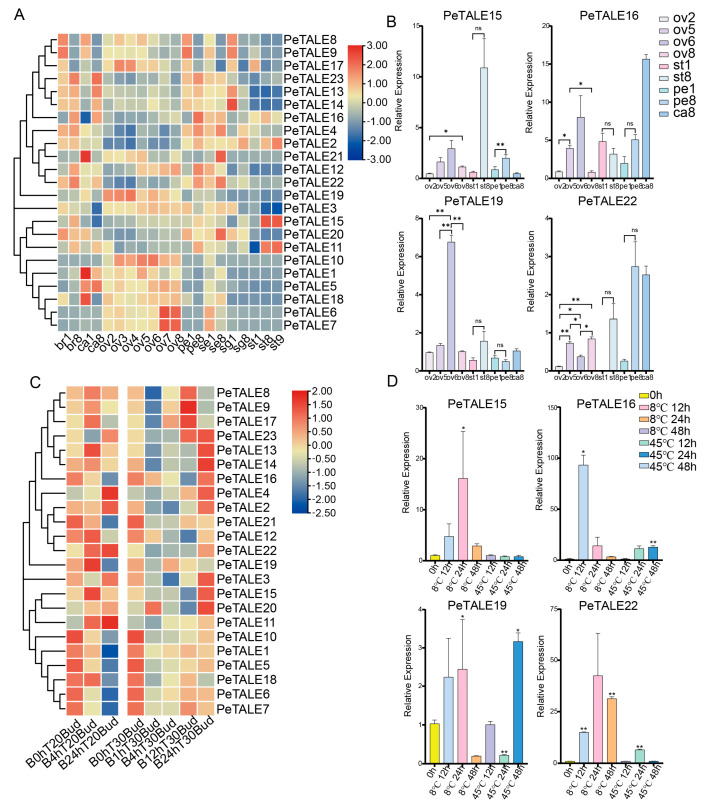 Figure 6
Figure 6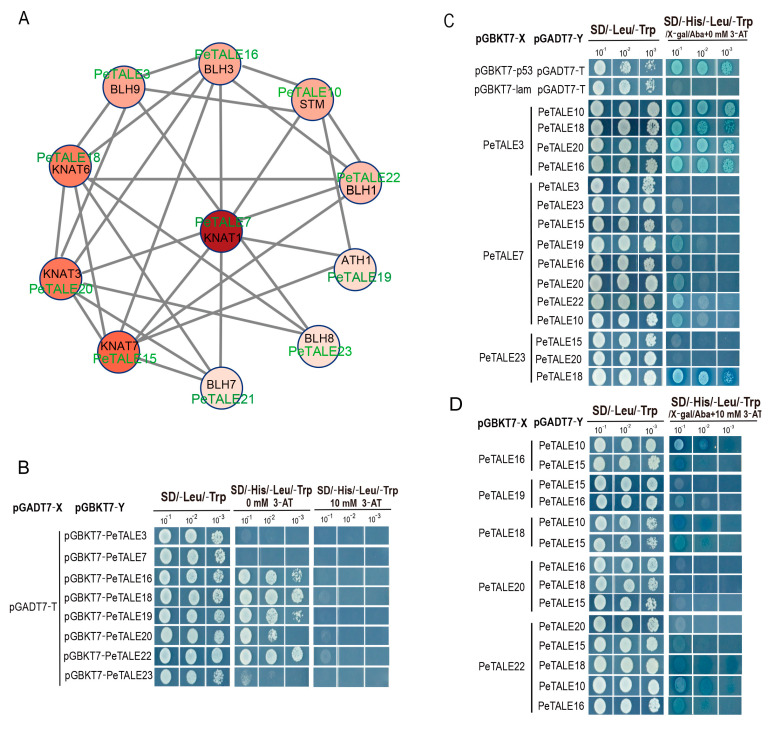 Figure 7
Figure 7- —Natural Science Foundation of Guangxi
- —National Natural Science Foundation of China
- —Science and Technology Major Project of Guangxi
- —Project of Guangxi
- —Major Science and Technology Project of Fujian Province
- —Guangxi Distinguished Experts Fellowship
- —Peking University
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsPlant Molecular Biology Research · Light effects on plants · Plant nutrient uptake and metabolism
1. Introduction
Plants maintain their lifelong organogenic capacity through stem cell reservoirs localized in meristematic tissues [1]. The shoot apical meristem (SAM), serving as the primary source for leaf and axillary meristem formation, undergoes a developmental transition into the inflorescence meristem during flowering initiation, ultimately generating floral meristems (FMs) that develop into gynoecia, which are thought to be modified leaves, with their margins defining lateral organ boundaries [2]. This developmental continuity between SAM dynamics and fruit patterning highlights conserved regulatory mechanisms governing plant morphogenesis [2].
Meristematic cell proliferation and differentiation are precisely orchestrated by transcription factor networks, integrating positional cues, differentiation states, and growth signals [1,2]. Central to this regulation are KNOTTED1-like homeodomain (KNOX) proteins, first characterized in maize [3]. In Arabidopsis, the functional interplay between SHOOT MERISTEMLESS (STM) and WUSCHEL (WUS) governs stem cell maintenance [4]. WUS sustains the central stem cell niche, while STM preserves SAM undifferentiation by modulating hormone homeostasis [2]. STM belongs to the Three-Amino-acid-Loop-Extension (TALE) homeodomain superclass of transcription factors, which includes both KNOTTED-like (KNAT or KNOX) and BEL1-like (BLH or BELL) proteins [5]. These TALE factors typically function as KNOX–BELL heterodimers, regulating a wide array of developmental processes, including meristem maintenance, leaf development, and flower formation [6,7]. Specifically, STM maintains the undifferentiated state of meristematic cells by repressing gibberellin (GA) biosynthesis, promoting GA catabolism, and stimulating cytokinin (CK) biosynthesis [8,9,10]. Furthermore, STM negatively regulates ASYMMETRIC LEAVES1 (AS1), a MYB transcription factor that antagonizes other TALE members (KNAT1/BREVIPEDICELLUS, KNAT2, and KNAT6) to regulate leaf patterning [2,11]. Lateral organ initiation requires the coordinated regulation of auxin and GA signaling alongside the downregulation of STM-related factors [7].
The TALE family, comprising KNOX and BELL subfamilies, features conserved structural domains: bipartite KNOX/BELL interaction domains upstream of the homeodomain, with KNOX proteins containing an ELK domain and BELL proteins possessing SKY and ZIBEL domains [6]. These proteins form functional heterodimers through KNOX–BELL domain interactions, regulating processes ranging from meristem maintenance to stress adaptation [12,13]. KNOX members (e.g., STM) predominantly govern meristem activity and organ patterning, whereas BELL proteins specialize in floral development and organ differentiation [12,13]. Mechanistically, TALE factors coordinate hormone signaling pathways, tuber formation, and stress response [14,15,16,17,18,19]. In Arabidopsis, KNOX genes are predominantly expressed in meristematic regions, regulating key processes such as cytokinin biosynthesis essential for maintaining SAM activity [10]. KNOX proteins are also implicated in secondary cell wall biosynthesis in poplar [20]. Additionally, KNOX and BELL genes play significant roles in regulating hormone pathways, such as those for gibberellins (GAs), abscisic acid (ABA), and cytokinin (CK), which are vital for plant growth and stress responses [21]. For example, the AtBLH1 and AtKNAT3 proteins form heterodimers to regulate seed germination through ABA signaling [22]. KNOX genes in A. thaliana influence CK biosynthetic gene expression to maintain SAM development [10], while in apple (Malus domestica), KNOX genes activate the ABA signaling pathway, promoting callus formation [23]. In response to abiotic stress, TALE gene expression is modulated by environmental stimuli. For example, soybean TALE gene promoters contain cis-elements responsive to salt and drought stresses [24]. Similarly, the overexpression of the KNOX-like gene TaKNOX11a from Triticum aestivum in A. thaliana enhances stress tolerance by modulating proline content and reducing oxidative damage [24]. These findings highlight that TALE genes regulate not only developmental processes but also plant adaption to environmental stresses.
Floral initiation, a critical step in the transition from vegetative to reproductive growth, is governed by a complex network of genetic regulators that integrate endogenous and environmental cues [25,26]. Temperature, in particular, plays a key role in floral induction and organ differentiation [27,28]. Plants must accurately interpret temperature signals over various timescales to synchronize development with seasonal changes. Although the mechanisms underlying acute thermal stress responses are relatively well-studied [29,30], the processes by which plants integrate long-term temperature signals to regulate floral development remain less understood. Passion fruit (Passiflora edulis), a tropical crop valued for its nutritional and economic significance [31], offers a unique system for studying temperature-sensitive floral organogenesis. Introduced to southern China in 1901, it achieves yields of up to 15 tons per hectare annually [32], yet exhibits notable thermal sensitivity: flower bud abortion at high temperatures (30/25 °C) and suppressed bud formation at cooler temperatures (20/15 °C) [33,34,35]. Its complex floral architecture (sepals, petals, corona, stamens, and carpels) makes it an excellent model for developmental studies.
Despite these attributes, the role of the floral TALE gene family in the thermal regulation of floral development in P. edulis remains unexplored. In this study, we conducted the first genome-wide analysis of PeTALE genes, combining RNA-seq profiling of temperature-stressed buds with structural, phylogenetic, and promoter analyses. Given their established roles in meristem identity, floral organogenesis, and hormone signaling, we focused on the KNOX and BELL subfamilies for functional analysis. We systematically investigated their chromosomal distribution, predicted protein interaction networks, and associations with hormone-responsive pathways to elucidate their functions in temperature-modulated floral development. This work provides a molecular framework for understanding thermal adaptation mechanisms in passion fruit and offers critical insights for breeding climate-resilient varieties.
2. Results
2.1. Identification of the TALE Gene Family in Passion Fruit
To identify candidate TALE members in P. edulis, we conducted a comprehensive search using the HMMER profile of TALE proteins as queries against the passion fruit protein database via the BLASTP algorithm. The resulting PeTALE candidate genes were further validated using the Pfam and SMART databases to confirm the presence of the characteristically conserved domains, including the homeodomain (HD) and KNOX or BEL1-like domains. Ultimately, a total of 23 PeTALE genes were identified and designated PeTALE1 to PeTALE23 based on their chromosomal positions (Table 1). Notably, the majority of these genes (10 out of 23) were located on chromosome LG01, while only a single gene was found on chromosome LG05, suggesting potential clustering of TALE family members on specific chromosomes (Figure S1). This clustering pattern may indicate gene duplication events or the existence of conserved genomic regions harboring multiple TALE genes, which could be linked to the regulation of key developmental processes. For a detailed analysis of the physical and biochemical properties of the PeTALE proteins, several key parameters were calculated, including protein length, molecular weight (MW), isoelectric point (pI), aliphatic index (A.I.), and predicted subcellular localization (Table 1). PeTALE protein lengths exhibited considerable variation, ranging from 195 amino acids (PeTALE11) to 811 amino acids (PeTALE21), reflecting functional diversity within the family. Correspondingly, molecular weight ranged from 22.506 kDa (PeTALE11) to 89.581 kDa (PeTALE21). The predicted isoelectric points varied from 4.70 (PeTALE5) to 8.75 (PeTALE22), indicating variability in charge properties across family members. We also assessed protein stability and hydrophobicity. According to the instability index [36], PeTALE1 was classified as a stable protein (instability index < 40), whereas the remaining members were predicted to be unstable. The aliphatic index (A.I.), which correlates with thermal stability, ranged from 60.12 (PeTALE6) to 83.99 (PeTALE15), suggesting that some PeTALE proteins may exhibit greater thermal stability than others. Furthermore, the grand average of hydropathicity (GRAVY) values, which ranged from −0.923 (PeTALE11) to −0.385 (PeTALE19), indicated that all identified PeTALE proteins are hydrophilic, potentially influencing their interaction with other hydrophilic molecules or their nuclear localization. Subcellular localization predictions revealed that all PeTALE proteins are likely localized to the nucleus (Table 1), consistent with their putative roles as transcription factors regulating gene expression during critical stages of plant development.
2.2. Classification and Phylogenetic Relationships of PeTALEs
To explore the evolutionary relationships and classification of TALE proteins in P. edulis, we constructed a phylogenetic tree using the neighbor-joining (NJ) method. The analysis included 104 TALE proteins from three species—Vitis vinifera (21 proteins), Arabidopsis thaliana (33 proteins), and Oryza sativa (27 proteins)—along with the 23 PeTALE proteins identified in this study. The resulting tree revealed two major clades, corresponding to the KNOX and BELL subfamilies, which were further subdivided into KNOX-I to KNOX-IV and BELL-I to BELL-V, respectively, based on specific domain signatures. Among the 23 PeTALE genes, 14 were assigned to the BELL subfamily, characterized by the presence of both a POX domain and a homeodomain, while the remaining 9 PeTALE genes were categorized within the KNOX subfamily, defined by the presence of the KNOX1 or KNOX2 domains, an ELK motif, and a homeodomain. Within the BELL subfamily, the largest cluster was BELL-V, containing more than 20 TALE genes, whereas BELL-I had the fewest members. The intermediate groups, BELL-II, BELL-III, and BELL-IV, each harbored a moderate number of genes. In the KNOX subfamily, we observed a relatively even distribution across the subgroups: KNOX-I contained only two TALE members, while KNOX-II, KNOX-III, and KNOX-IV each included a larger number of TALE genes (Figure 1). The distribution of PeTALE genes across these subfamilies and subgroups reflects the evolutionary diversification of this gene family in P. edulis. Notably, the BELL subfamily, typically associated with floral development and organ identity regulation, appears to have undergone greater expansion in passion fruit compared to the KNOX subfamily, which is mainly linked to shoot and leaf development.
To assess the evolutionary pressure acting on the PeTALE genes, we calculated the non-synonymous (Ka) to synonymous (Ks) substitution ratio (Ka/Ks). This ratio provides insights into the selective forces shaping gene evolution: a Ka/Ks ratio less than 1 indicates purifying selection, a ratio equal to 1 suggests neutral selection, and a ratio greater than 1 is indicative of positive selection. Our analysis revealed that all PeTALE genes exhibited Ka/Ks ratios of less than 1 (Table 2), suggesting that these genes have predominantly undergone purifying selection throughout their evolutionary history.
2.3. Chromosomal Distribution and Synteny Analysis of PeTALE Genes
The chromosomal distribution of the 23 identified PeTALE genes was mapped based on the P. edulis genome sequences. The results revealed that the PeTALE genes are dispersed across multiple chromosomes, with each chromosome harboring one or more genes. Notably, chromosome 1 (LG01) contains the highest number of PeTALE genes, hosting 10 out of 23 (43.48%), while chromosome 5 (LG05) is home to only a single gene (4.35%). Chromosome 2 (LG02) contains 4 genes (17.39%), and chromosomes LG06, LG07, and LG08 each harbor 2 to 3 PeTALE genes. Interestingly, no PeTALE genes were located on chromosomes LG03, LG04, and LG09 (Figure S1), suggesting that gene loss or chromosomal rearrangements may have occurred during the evolution of the TALE gene family in P. edulis.
To explore the mechanisms driving the expansion of the PeTALE gene family, we conducted a synteny analysis to identify both intra- and inter-species duplications. Intra-species synteny analysis revealed 18 pairs of duplicated PeTALE genes, indicative of gene duplication events within the P. edulis genome. Notable gene pairs include PeTALE8/PeTALE9, PeTALE2/PeTALE4, PeTALE1/PeTALE5, PeTALE8/PeTALE13, PeTALE8/PeTALE14, PeTALE9/PeTALE13, PeTALE9/PeTALE14, PeTALE9/PeTALE16, PeTALE8/PeTALE17, PeTALE8/PeTALE23, PeTALE9/PeTALE23, PeTALE2/PeTALE22, PeTALE13/PeTALE14, PeTALE13/PeTALE16, PeTALE13/PeTALE23, PeTALE14/PeTALE23, PeTALE12/PeTALE22, and PeTALE16/PeTALE23 (Figure 2A; Table S1). Tandem duplicates were also evident, particularly in the PeTALE6/PeTALE7 and PeTALE16/PeTALE17 clusters, highlighting the role of local duplications in the expansion of this gene family (Figure S1). Inter-species synteny comparisons further revealed significant evolutionary relationships between P. edulis and other species. In particular, 26 syntenic gene pairs were identified between P. edulis and Vitis vinifera, such as PeTALE5/VvTALE9, PeTALE10/VvTALE16, PeTALE5/VvTALE2, PeTALE1/VvTALE2, PeTALE8/VvTALE5, PeTALE9/VvTALE5, PeTALE6/VvTALE3, PeTALE3/VvTALE18, PeTALE2/VvTALE7, PeTALE11/VvTALE8, PeTALE13/VvTALE5, PeTALE14/VvTALE5, PeTALE13/VvTALE4, PeTALE14/VvTALE14, PeTALE13/VvTALE14, PeTALE11/VvTALE20, PeTALE12/VvTALE21, PeTALE15/VvTALE13, PeTALE16/VvTALE5, PeTALE18/VvTALE17, PeTALE19/VvTALE19, PeTALE20/VvTALE20, PeTALE23/VvTALE5, PeTALE23/VvTALE14, PeTALE21/VvTALE15, and PeTALE22/VvTALE21 (Figure 2B; Table S2). Among these, PeTALE13 exhibited synteny with three distinct VvTALE genes (VvTALE5, VvTALE4, and VvTALE14), suggesting a complex pattern of gene conservation and divergence. A similar synteny analysis with Arabidopsis thaliana identified 32 gene pairs, including PeTALE8/AtTALE11, PeTALE5/AtTALE8, PeTALE1/AtTALE5, PeTALE5/AtTALE5, PeTALE10/AtTALE6, PeTALE1/AtTALE8, PeTALE9/AtTALE3, PeTALE2/AtTALE20, PeTALE4/AtTALE20, PeTALE3/AtTALE29, PeTALE15/AtTALE7, PeTALE14/AtTALE10, PeTALE14/AtTALE3, PeTALE13/AtTALE10, PeTALE13/AtTALE3, PeTALE14/AtTALE12, PeTALE13/AtTALE12, PeTALE14/AtTALE10, PeTALE13/AtTALE26, PeTALE14/AtTALE26, PeTALE23/AtTALE10, PeTALE23/AtTALE3, PeTALE23/AtTALE12, PeTALE22/AtTALE17, PeTALE23/AtTALE26, PeTALE20/AtTALE23, PeTALE19/AtTALE24, PeTALE20/AtTALE30, PeTALE20/AtTALE31, PeTALE16/AtTALE10, PeTALE16/AtTALE3, and PeTALE16/AtTALE26 (Figure 2B; Table S2). These results underscore the evolutionary relationships between PeTALE genes in passion fruit and those in distantly related species like grapevine and Arabidopsis, pointing to shared ancestral origins and functional conservation across these plant species. These synteny analyses not only highlight the presence of both tandem and segmental duplications within the PeTALE gene family but also provide insights into the evolutionary processes shaping the expansion and diversification of TALE genes in P. edulis. The conservation of PeTALE gene pairs across species further suggests that these genes have maintained crucial developmental functions over evolutionary time.
2.4. Conserved Motifs and Gene Structure Characteristic Analysis of PeTALE Genes
The structural characteristics of PeTALE genes were examined by analyzing the conserved motifs and intron–exon organization of their coding sequences using the GFF annotation file from the P. edulis genome (Figure 3). A total of ten conserved motifs were identified across the PeTALE family. The core TALE domain, essential for the functional properties of most PeTALE proteins, was characterized by the presence of Motifs 1, 2, 9, and 10 (Figure 3B). However, certain motifs were either absent or uniquely present in specific PeTALE proteins. For example, PeTALE11 was composed solely of Motifs 1, 2, and 10, while Motif 9 was missing in PeTALE8, PeTALE9, PeTALE11, PeTALE17, and PeTALE20. These variations in motif composition suggest that different PeTALE proteins may fulfill distinct biological functions, potentially modulated by specific motifs in response to particular cellular or environmental cues.
In addition to motif conservation, our analysis revealed that genes within the same subfamily often share similar structure features. For instance, five PeTALE genes (PeTALE8, PeTALE9, PeTALE13, PeTALE14, and PeTALE17) each harbor three introns, whereas PeTALE2 and PeTALE21 possess four introns, and PeTALE15 and PeTALE20 are characterized by six introns (Figure 3C). Furthermore, 12 PeTALE genes (PeTALE1, PeTALE3, PeTALE6, PeTALE10, PeTALE11, PeTALE12, PeTALE14, PeTALE15, PeTALE18, PeTALE19, PeTALE20, and PeTALE23) contain both 5′ and 3′-UTRs, while 8 genes (PeTALE2, PeTALE4, PeTALE5, PeTALE7, PeTALE8, PeTALE9, PeTALE13, and PeTALE22) lacked a 5′-UTR, and 3 genes (PeTALE16, PeTALE17, and PeTALE21) lacked a 3′-UTR (Figure 3C). Notably, gene structure tended to be highly conserved within the same clade, while greater variation was observed between clades. For instance, genes such as PeTALE8, PeTALE9, PeTALE13, PeTALE14, and PeTALE17, which share a common intron–exon structure pattern (three introns), were clustered together (Figure 3C), suggesting a potential link between gene structure and evolutionary lineage.
To further investigate the potential regulatory mechanisms of PeTALE genes, we examined the cis-elements within their promoter regions, defined as the 2000 bp sequence upstream of the transcriptional start codon. This analysis revealed a diverse array of cis-elements, including those associated with plant growth (9 elements), stress responses (6 elements), light signaling (22 elements), as well as unique elements such as an AT-rich DNA-binding protein (ATBP-1) binding site and an A-box element (Figure 4). Most PeTALE genes (except PeTALE21 and PeTALE22) contained at least one light-responsive cis-element in their promoter regions, indicating a potential role in light-mediated gene regulation. Additionally, the majority of PeTALE genes (with the exceptions of PeTALE21, PeTALE15, PeTALE11, PeTALE23, and PeTALE22) contained an MeJA-responsive cis-acting element, suggesting that these genes may participate in phytohormone signaling pathways, particularly in response to MeJA, which is crucial for plant defense mechanisms and stress responses.
2.5. The Secondary Structure Prediction of PeTALE Proteins
Understanding protein structure is fundamental for elucidating its functional roles and cellular localization. Three-dimension (3D) structural models of PeTALE proteins from different subfamilies were generated using the SWISS-MODEL platform, and the quality of each model was evaluated based on the GMQE and Q-mean scores. These quality metrics were used to select the most reliable models for further analysis. The results revealed that, while there was notable variability in the 3D structures of PeTALE proteins across subfamilies, proteins within the same subfamily displayed highly conserved spatial configurations (Figure 5A). For instance, PeTALE8 and PeTALE9, PeTALE2 and PeTALE19, and PeTALE22 and PeTALE23 exhibited similar secondary structure arrangements, supporting the hypothesis that proteins within closely related groups share common structural features (Figure 5A).
The structural diversity among these proteins is strongly influenced by their secondary structure composition. To explore this further, we conducted a secondary structure prediction analysis for the PeTALE proteins. The results revealed that the majority of PeTALE proteins are predominantly composed of alpha helices, extended strands, and random coils, with the respective proportions ranging from 23.18% to 50.77%, 0.65% to 3.48%, and 45.82% to 73.74%, respectively (Figure 5B). Interestingly, members of the KNOX subfamily, such as PeTALE1 and PeTALE15, exhibited distinct structural profiles characterized by a complete absence of extended strands and a substantial increase in random coil content, averaging 62.39%. In contrast, the alpha helix structure remained the most prevalent overall, accounting for an average of 35.72% of the total sequence, while extended strands represented only about 2.07% (Figure 5B).
2.6. Expression Profiling of PeTALE Genes For Different Floral Organs and Their Response to Temperature
To gain insight into the potential roles of the 23 PeTALE genes, their expression patterns were analyzed using RNA-seq data from various tissues and developmental stages in P. edulis. Expression levels were assessed across a range of tissues, including bracts (br1 and br8), coronas (Ca1 and Ca8), ovules (Ov S2-S8), petals (Pe1 and Pe8), sepals (Se1 and Se8), stigmas (Sg1 and Sg8), and stamens (St1, St8, and St9). Distinct expression profiles were observed across the different PeTALE subfamilies. Genes from the BELL subfamily exhibited diverse expression patterns. For instance, PeTALE8, PeTALE9, PeTALE13, and PeTALE14 (BELL-II group) were highly expressed during the early developmental stages, whereas PeTALE2 and PeTALE4 (also BELL-II group) showed enhanced expression during the mid- and late stages of stigma development (Figure 6A), suggesting their potential involvement in stigma morphogenesis. Several BELL genes, such as PeTALE13 and PeTALE14, were strongly expressed during early stigma development, as well as in petals and during the final stages of corona development. Similarly, PeTALE8 and PeTALE9 exhibited elevated expression during early stages of petal, corona, and stigma development and throughout most stages of ovule development. Notably, PeTALE21 showed strong, specific expression during the early stages of corona development. Within the BELL subfamily, genes such as PeTALE23, PeTALE16, PeTALE4, and PeTAL22 displayed preferential expression in sepals, with slightly higher levels also observed in petals compared to other tissues. PeTALE17 was notably expressed at high levels during early petal, stigma, stamen, and ovule development (Figure 6B). In contrast, PeTALE4 showed strong expression during the late stages of corona and stamen development, with moderate expression throughout the petal and sepal stages. Although PeTALE22 exhibited relatively lower expression in stamens, stigmas, and ovules, it demonstrated higher expression during certain stages of corona development, aligning with the expression profile of PeTALE4. The expression of some PeTALE genes was highly tissue-specific. For instance, PeTALE19 and PeTALE3 were predominantly expressed across ovule development stages, while PeTALE11 and PeTALE15 were highly expressed only during later stages of stamen development. Moreover, the expression levels of several genes exhibited temporal variation. For example, PeTALE13, PeTALE14, and PeTALE16 showed progressively increasing expression during corona and petal development, whereas PeTALE22 exhibited a similar upward trend specifically in sepals (Figure 6B).
To assess the temperature sensitivity of PeTALE genes, RNA-seq data were analyzed from flower buds exposed to cold (20 °C) and heat (30 °C) stress across different time points (Figure 6C). After excluding genes with low expression, notable differences emerged under temperature stress. Cold stress led to the upregulation of genes such as PeTALE11, PeTALE20, PeTALE3, PeTALE22, PeTALE2, PeTALE23, and PeTALE4, with PeTALE11, PeTALE3, PeTALE4, and PeTALE23 exhibiting significant increases as treatment duration increased (Figure 6C). Conversely, heat stress responses were more nuanced. While most genes initially showed elevated expression, some (PeTALE8, PeTALE9, PeTALE17, and PeTALE19) were upregulated at the 1 and 4 h time points but downregulated after 12 and 24 h. Additionally, genes such as PeTALE23, PeTALE13, PeTALE4, PeTALE22, PeTALE15, and PeTALE20 peaked at specific times, while PeTALE10, PeTALE18, and PeTALE21 were repressed under heat conditions (Figure 6D). These findings suggest that specific PeTALE subfamilies exhibit distinct temperature-responsive behaviors: BELL-II genes (e.g., PeTALE3) and BELL-IV genes (e.g., PeTALE4) primarily respond to cold stress, whereas KNOX-IV (PeTALE10), KNOX-III (PeTALE18), and certain BELL-II genes (e.g., PeTALE21) are more responsive to heat stress. Together, these results offer valuable insights into the roles of PeTALE genes in developmental regulation and stress responses in P. edulis, providing a foundation for future functional studies on plant growth and adaptation.
2.7. Protein Interaction of PeTALE Proteins
To further investigate the functional conservation of the PeTALE gene family, we performed a protein–protein interaction (PPI) analysis to predict potential interacting partners of PeTALE proteins (Figure 7A). The analysis revealed that PeTALE7, an ortholog of AtKNAT1, exhibited high expression in ovules and formed a robust interaction network with multiple PeTALE proteins, based on their Arabidopsis orthologs (including STM, ATH1, BLH1/3/7/8/9, and KNAT3/6/7). This interaction cluster (comprising PeTALE7/8/9/10/15/16/18/19/20/21/22/23) suggests a conserved role for PeTALE7 and its associated genes in floral organogenesis and reproductive tissue development (Figure 7A). Additionally, we identified 10 orthologous proteins (STM, ATH1, BLH1/3/7/8/9, and KNAT3/6/7) as candidate interactors of PeTALE3/7/16/18/19/20/22/23, with varying interaction strengths observed among them (Figure 7A).
To validate these interactions, yeast two-hybrid (Y2H) assays were conducted. Initial transcriptional activity screening revealed that all transformed yeast strains grew normally on SD/-Leu-Trp medium (Figure 7B). Five co-transformed yeast strains (pGBKT7-PeTALE16/18/19/20/22) exhibited growth on SD/-His-Leu-Trp medium (0 mM 3-AT) and displayed blue coloration on X-α-Gal-supplemented SD/-His-Leu-Trp medium (0 mM 3-AT), confirming their transcriptional activation capacity. However, growth was suppressed on SD/-His-Leu-Trp medium supplemented with 10 mM 3-AT, indicating transcriptional inhibition (Figure 7B). In contrast, three plasmids (pGBKT7-PeTALE3/7/23) showed no growth or coloration under the same conditions, confirming their lack of transcriptional activation. Subsequent Y2H assays for 29 potential KNOX–BELL combinations demonstrated direct interactions between PeTALE3 and three KNOX proteins (PeTALE10/18/20), as well as one BELL protein (PeTALE16), while PeTALE18 interacted with two BELL proteins (PeTALE23 and PeTALE22) (Figure 7C,D). Weak interactions were observed between PeTALE7 and PeTALE19/20/22/10, as well as among PeTALE10–PeTALE16, PeTALE10–PeTALE18, and PeTALE10–PeTALE22 combinations (Figure 7C,D). These findings highlight a complex interaction network among PeTALE proteins, emphasizing their potential regulatory roles in floral organogenesis and reproductive tissue development.
3. Discussion
3.1. Evolutionary Conservation and Diversification of PeTALE Genes
As a globally significant horticultural crop, Passiflora edulis offers unique advantages for studying floral organogenesis due to its intricate flower morphology and temperature-sensitive reproductive development [34]. The TALE gene family is widely distributed across plant genomes and plays a crucial role in regulating various aspects of plant growth, development, and stress responses. Previous genome-wide analyses of TALE genes have been conducted in numerous species, including Arabidopsis [6], Populus [20], Punica granatum [35], and Glycine max [36], highlighting considerable variability in TALE gene numbers across different species [20,35]. For instance, there are 103 TALE genes in Switchgrass, 33 in Arabidopsis, 27 in Oryza sativa, and only 18 in Ananas comosus. TALE genes regulate diverse physiological processes, including fruit development in Solanum lycopersicum [37] and stress responses in Cucumis sativus [38] and Triticum aestivum [24]. In this study, our genome-wide analysis identified 23 PeTALE genes—comparable to Prunus armeniaca (22 genes) but slightly higher than Vitis vinifera (21 genes)—with predicted nuclear localization (Table 1) and conserved chromosomal distribution patterns (Figure S1). The identified PeTALE genes are conserved in other Passiflora species with varying floral morphologies. Based on the well-established roles of KNOX and BELL genes in meristem identity, floral organogenesis, and hormone responses, it is likely that functional conservation exists across the Passiflora genus, due to the high sequence similarity observed. This point has been incorporated into phylogenomic studies across the Passiflora genus to better understand the link between gene diversification and floral morphology. Phylogenetic classification resolved these genes into nine clades (four KNOX and five BELL), mirroring evolutionary patterns observed in dicots, where KNAT subgroups are retained but lost in monocots (Figure 1). Structural conservation of the homeodomain motifs (Homeobox-KN, KNOX1/2, and ELK) further supports their functional roles in DNA-binding and transcriptional regulation [6]. Specifically, the C-terminal homeobox-KN domain facilitates DNA interaction, while the ELK domain directs nuclear localization [12,22,39,40]. These findings corroborate the functional conservation of TALE proteins across angiosperms, while also highlighting lineage-specific adaptations in P. edulis.
3.2. PeTALEs in Floral Organogenesis: From Meristem Dynamics to Organ-Specific Regulation
Synteny analysis identified 18 collinear PeTALE pairs, predominantly within the BELL subfamily, with tandem duplication events contributing significantly to TALE gene expansion. The expansion of BELL genes may be associated with the evolutionary adaptation of Passiflora to its unique reproductive strategy and complex floral structures. Comparative genomics suggest that lineage-specific duplications of BELL genes could underlie the developmental specialization necessary for the elaborate coronas, ovules, and stigmas observed in Passiflora. This dominance likely reflects an evolutionary bias favoring BELL-mediated transcriptional regulation during floral organ identity specification. The expansion of BELL genes appears tightly linked to the increased floral complexity of Passiflora. BELL proteins are known to form heterodimers with KNOX proteins to regulate developmental boundaries and meristem maintenance. In Passiflora, this interaction may have been co-opted to refine the differentiation of additional floral whorls, thereby contributing to the species’ intricate floral structures. This suggests a functional co-evolution between PeBELL genes and floral architectural innovations.
Purifying selection (Ka/Ks < 1) likely preserved the core developmental functions of these genes during evolution. Inter-species comparisons revealed stronger syntenic conservation with Arabidopsis than with monocots, supporting conserved regulatory roles in dicot development. Gene structure analysis demonstrated conserved intron–exon arrangements within the PeTALE family, consistent with findings in other studies such as Populus [20]. Moreover, genomic architecture analysis showed that all identified PeTALE genes possess at least one intron, with highly conserved structural features across family members. Protein motif analysis confirmed that members within the same subfamily share identical motifs, reinforcing the notion of functional conservation [41]. Promoter profiling uncovered abundant cis-elements linked to light responsiveness (45.83%), phytohormone signaling (18.75%), and stress adaptation (18.75%), including low-temperature-responsive elements (LTRs) critical for cold stress regulation. This aligns with P. edulis’s sensitivity to temperature fluctuations [20,34], suggesting that PeTALEs may orchestrate transcriptional responses to temperature fluctuations via LTR-mediated pathways.
3.3. Temperature-Responsive PeTALEs: Balancing Development and Stress Adaptation
Expression profiling across floral developmental stages revealed that PeTALE genes exhibit clear functional specialization. During early organogenesis, PeTALE8/9 showed peak expression during the initiation of petals, coronas, stigmas, and ovules, paralleling the roles reported for BEL1 homologs in ovule development [40,42]. Tissue-specific regulation was evident, with PeTALE21 displaying strong expression during the early stages of corona development, suggesting a critical role in corona formation. Similarly, PeTALE23/16/4/22 showed preferential expression in sepals, echoing findings from Populus where TALE factors are involved in organ patterning [20]. In terms of maturation control, PeTALE13/14/16 exhibited progressive upregulation during petal and corona development, suggesting their involvement in the maturation of these floral organs. Recent studies have further highlighted the roles of TALE genes in reproductive development: in pomegranate, TALE genes were predicted to regulate SAM, flower, and ovule development [35], while in walnut, TALE gene expression varied across flower bud developmental stages [43]. In cotton, the downregulation of the TALE family member GhSTM3 affected flowering time [44]. While KNOX1 genes such as STM and KNAT1 in Arabidopsis are largely restricted to meristematic tissues, certain PeTALE genes (PeBELL7 and PeKNOX3) exhibit expression in floral organs such as the ovary and corona, suggesting a divergence in spatial regulation. This may reflect neofunctionalization events post-duplication that allowed PeTALE genes to acquire novel roles in reproductive development.
Temperature stress significantly modulated the expression of several PeTALE genes. For instance, the genes PeTALE3/4 (BELL subfamily) were upregulated under cold stress, while PeTALE10/18 (KNOX subfamily) responded preferentially to heat stress. These findings highlight the potential roles of the PeTALE genes in mediating plant response to environmental stress. The cold-induced expression of PeTALE3/4 correlates with the presence of LTR cis-elements in their promoters, suggesting a mechanism for transcriptional reprogramming under chilling stress—a key adaptive trait for P. edulis, which is highly sensitive to bud abortion under cold temperatures [33,34]. Conversely, the heat-activated expression of PeTALE10/18 may help counteract thermal damage, potentially through mechanisms similar to wheat TaKNOX11a’s role in mitigating oxidative stress [45]. This functional dichotomy mirrors findings in Gossypium and Prunus, where TALE genes differentially regulate responses to abiotic stresses such as drought and salt stress [37,45,46], further supporting their pleiotropic roles in stress adaptation. We hypothesize that BELL genes like PeTALE3 and PeTALE4 mediate cold stress response in P. edulis, enhancing flower bud resilience under low temperatures. Moreover, temperature-sensitive PeTALEs may act as molecular hubs, integrating environmental signals with hormonal pathways. For example, PeBELL2 and PeKNOX5, which are downregulated under elevated temperatures, may modulate GA biosynthesis repressors or interact with ABA-responsive promoter elements, thereby influencing flowering time and organ differentiation under fluctuating thermal conditions.
In conclusion, this study provides a comprehensive analysis of the PeTALE gene family in Passiflora edulis, shedding light on their roles in floral organ development and stress adaptation. However, further functional studies are necessary to fully elucidate the regulatory networks and molecular mechanisms underlying these processes. Such investigations will be crucial for understanding how P. edulis adapts to environmental challenges and how TALE genes orchestrate complex developmental programs, ultimately informing future crop improvement strategies.
4. Materials and Methods
4.1. Identification of Putative TALE Gene Members in P. edulis
The P. edulis genome and proteome datasets were obtained from the National Genomics Data Center (NGDC, accession number GWHAZTM00000000, https://ngdc.cncb.ac.cn/ (accessed on 25 September 2024)) [47]. To identify putative TALE gene members within the P. edulis genome, we employed two complementary approaches: Hidden Markov Model (HMM) searching and BLAST-based sequence alignment. For the HMM approach, the TALE family domain (PF00046) from the Pfam database (www.pfam.org (accessed on 25 September 2024)) was used to scan the P. edulis protein sequences using HMMER3 software (v3.4, http://hmmer.janelia.org/ (accessed on 25 September 2024)) [48]. Simultaneously, a BLASTP v2.12.0 search was conducted using known plant TALE protein sequences retrieved from the NCBI database as query sequences. Sequences with an E-value threshold of <1 × 10^−5^ were retained for further analysis. Non-redundant candidates from both methods were consolidated and subsequently subjected to domain validation using SMART (http://smart.embl-heidelberg.de (accessed on 27 September 2024)) [49] and the Conserved Domain Database (CDD, www.ncbi.nlm.nih.gov/cdd (accessed on 28 September 2024)) [50]. Only sequences containing both a homeodomain (HD) and either KNOX1/KNOX2 (for the KNOX subfamily) or a BELL/POX (for the BELL subfamily) gene were retained. Based on domain composition, the identified genes were classified into two major subfamilies: the KNOX subfamily, characterized by the presence of HD and KNOX1/KNOX2 domains, and the BELL subfamily, characterized by HD and BELL/POX domains. In total, 23 TALE genes were identified and named according to their chromosomal positions in the P. edulis genome for consistency. To predict their potential cellular roles, subcellular localization was assessed using WoLF PSORT (https://wolfpsort.hgc.jp (accessed on 16 November 2024)). Additionally, the key physicochemical properties of the encoded proteins, including the molecular weight (MW), isoelectric point (pI), and grand average of hydropathicity (GRAVY), were calculated using the ExPASy v3.0 online tools [51].
4.2. Sequence Alignment and Phylogenetic Analyses
To elucidate the evolutionary relationships of TALE genes, a comparative phylogenetic analysis was conducted across multiple plant species. Full-length coding sequences of P. edulis TALE genes were extracted using TBtools-II (v 2.119) [52]. TALE protein sequences from other plant species, including Oryza sativa, Vitis vinifera, and Arabidopsis thaliana, were retrieved from the PlantTFDB v4.0 database (http://planttfdb.gao-lab.org/ (accessed on 16 December 2024)). For sequence alignment, multiple sequence alignments of the TALE proteins were performed using the MUSCLE algorithm implemented in MEGA 11 [53], with manual curation to optimize the representation of conserved domains. Based on the aligned sequences, a phylogenetic tree was constructed using the neighbor-joining (NJ) method in MEGA 11, with bootstrap support values calculated from 1000 replicates to assess the reliability of the tree topology. The resulting phylogenetic tree was subsequently annotated and visualized using the iTOL web tool (https://itol.embl.de (accessed on 30 January 2025)) [54].
4.3. Chromosomal Localization and Duplication Analysis of TALE Genes
The chromosomal locations of TALE genes in P. edulis were identified using the passion fruit genome annotation file, and the genes were subsequently mapped to their respective chromosomes. Visualization of chromosomal localization was performed using TBtools software (v 2.119) [52], providing a clear representation of the distribution of TALE genes across the genome. To investigate the evolutionary dynamics of the P. edulis genome, particularly regarding gene duplication events, a gene duplication analysis was conducted using the Multiple Collinearity Scanning Toolkit (MCScanX) [55] with default settings. Genomic data for Oryza sativa, Arabidopsis thaliana, and Vitis vinifera were retrieved from the JGI Phytozome database (https://phytozome-next.jgi.doe.gov (accessed on 15 February 2025)) for comparative analysis. A comparative synteny approach was employed to assess orthologous relationships and gene duplication events between TALE genes in P. edulis and these three reference species. Syntenic blocks of TALE superfamily members across species were visualized using TBtools, highlighting conserved chromosomal regions and identifying potential evolutionary conserved gene clusters.
To further understand the selective pressures acting on TALE genes, Ka (non-synonymous) and Ks (synonymous) substitution rates were calculated for tandem and segmental duplication events using TBtools [52]. Selection modes were inferred based on Ka/Ks ratios: Ka/Ks > 1 indicated positive selection, Ka/Ks < 1 suggested purifying selection, and Ka/Ks = 1 reflected neutral evolution. Furthermore, the divergence times of segmental duplications were estimated using the formula T = Ks/(2λ), assuming a synonymous substitution rate (λ) of 6.5 × 10^−9^ substitutions per site per year [52].
4.4. Conserved Motif Prediction and Cis-Acting Element Analysis
Conserved protein motifs within the TALE members were identified using the MEME suite (https://meme-suite.org/meme/ (accessed on 23 February 2025)) [56], with the following parameters: maximum number of motifs set to 10, and motif width ranging from 6 to 50 residues. Exon–intron structures of the TALE genes were determined by aligning their coding sequences to the corresponding genomic DNA sequences using the genome annotation GFF3 files. The conserved regions within the TALE domains were further verified through the CDD database (www.ncbi.nlm.nih.gov/cdd (accessed on 23 February 2025)) [50].
To explore potential regulatory elements governing the expression of TALE genes, 2000 bp genomic sequences upstream of each gene’s translation start site were extracted as putative promoter regions. Cis-regulatory elements within these regions were predicted using PlantCARE (http://bioinformatics.psb.ugent.be/webtools/plantcare/html (accessed on 27 February 2025)) [57], followed by manual curation to annotate elements associated with stress responses, hormonal signaling, and developmental regulation.
4.5. Tertiary Structure Analysis of TALE Family Member
To investigate the three-dimensional (tertiary) structures of TALE proteins, all protein sequences were submitted to the SWISS-MODEL server (https://swissmodel.expasy.org (accessed on 8 March 2025)) [58] using a homology-based modeling approach, with template selection guided by the Protein Data Bank (PDB, www.rcsb.org (accessed on 12 March 2025)). Secondary structural elements, including alpha-helices, beta-sheets, and loops, were annotated based on alignments with the selected reference models. After the structural models were generated, the resulting three-dimensional configurations were refined and visualized using PyMOL v3.1 [59] to analyze domain architecture, protein fold patterns, and potential functional motifs.
4.6. Plant Material and Sample Preparation
Healthy two-month-old P. edulis (cultivar Qinmi) plants were cultivated in controlled-climate chambers at the Institute of Horticulture, Guangxi Academy of Agricultural Sciences, China. Temperature stress treatments were conducted following the protocol outlined by An et al. (2024) [47]. For cold stress, plants were transferred to a growth chamber set to 20 °C, while for heat stress, they were exposed to 30 °C. Floral buds were sampled at various time intervals to capture the stress response: 12, 24, and 48 h for cold and heat stresses. Control plants were maintained at 25 °C throughout the experiment. Three biological replicates were performed. Floral buds were collected at the designated time points, immediately frozen in liquid nitrogen, and stored at −80 °C. Total RNA was extracted from 100 mg samples using the RNAprep Pure Plant Kit (Tiangen Biotech (Beijing) Co., Ltd., Beijing, China) according to the manufacturer’s protocol. RNA quality and concentration were assessed using NanoDrop 2000 spectrophotometry (Thermo Fisher, Waltham, MA, USA) (A260/A280 > 1.8) and confirmed by agarose gel electrophoresis.
4.7. RNA Isolation, qRT-PCR, and Expression Pattern Analysis
Total RNA was isolated from P. edulis tissues using the RNAprep Pure Plant Kit (Tiangen Biotech) according to the manufacturer’s instructions. RNA concentrations were determined, and 1 µg of RNA per biological replicate was used for PCR library construction. Sequencing libraries were prepared using the NEBNext Ultra RNA Library Prep Kit (NEB, Beverly City, MA, USA) for Illumina platforms. RNA integrity was verified using a NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific) and confirmed by agarose gel electrophoresis. The expression levels of PeTALE genes were assessed by synthesizing cDNA from total RNA using the ThermoScript RT-PCR kit (Thermo Fisher Scientific, Carlsbad, CA, USA). qRT-PCR was conducted to analyze gene expression across various floral developmental stages, including Bracts1−8 (br1−br8), Sepals 1−8 (Se1−Se8), Petals 1−8 (Pe1−Pe8), Corona1−8 (Ca1p−Ca8), Stamens1,8,9 (St1, St8 and St9), Stigma1−8 (Sg1−Sg8), and Ovules 2−8 (Ov2−Ov8). All samples were collected and immediately frozen in liquid nitrogen for subsequent RNA extraction. Bar graphs were generated using GraphPad Prism 10.1.2 software. Relative gene expression was calculated using the 2^−∆∆Ct^ method, with statistical significance indicated by * (p ≤ 0.05) and ** (p ≤ 0.01), as determined by Student’s t-test.
Four PeTALE genes (PeTALE15, 16, 19, and 22) from the KNOX-II, BELL-II, BELL-I, and BELL-IV clusters, respectively, were selected for qRT-PCR validation based on their notable expression profiles identified in the RNA-seq analysis. These genes exhibited distinct expression patterns in reproductive organs or under temperature treatments, suggesting potential involvement in floral morphogenesis and stress-responsive regulatory pathways. For qRT-PCR, reactions were performed under the following conditions: initial denaturation at 95 °C for 30 s, followed by 40 cycles of 95 °C for 10 s and 60 °C for 30 s. Each 20 µL reaction contained 1 µL of cDNA template, 10 µL of 2 × Taq Pro Universal SYBR qPCR Master Mix (Vazyme, Nanjing, China), 0.4 µL of each primer (10 µM), and 8.2 µL of ddH_2_O. Amplifications were conducted using a Bio-Rad Real-time PCR system (Foster City, CA, USA), with three biological replicates per sample. EF1α was used as the reference gene for normalization [47].
For RNA-seq analysis, raw reads were pre-processed to obtain clean reads using Trimmomatic software v3.06 with default settings. Cleaned reads were subsequently mapped to the P. edulis reference genome using Hisat2 software v2.2.1. Gene expression levels were quantified by calculating FPKM (fragments per kilobase of transcript per million mapped reads) values using Cufflinks. Log2-transformed FPKM values were used to determine the expression patterns of PeTALE genes, and a heatmap was generated using TBtools software [52].
4.8. Protein–Protein Interaction (PPI) Network Construction of PeTALEs
To predict potential protein–protein interactions (PPIs) among P. edulis TALE proteins, we utilized the Search Tool for the Retrieval of Interacting Genes/Proteins (STRING) database (http://cn.string-db.org/ (accessed on 15 March 2025)), a well-established platform for exploring molecular interactions. Candidate PPI pairs were identified using the STRING database, with a combined confidence score greater than 0.4, reflecting a moderate level of interaction confidence. The resulting PPI data were visualized and analyzed by constructing an interaction network using Cytoscape (v3.8.2, https://cytoscape.org/ (accessed on 15 March 2025)), a powerful tool for network analysis and visualization. This approach enabled us to map the interaction relationships among PeTALE proteins and infer their potential functional roles in cellular processes based on network connectivity patterns.
4.9. Yeast Two-Hybrid Assay
To validate the predicted interactions, we employed the yeast two-hybrid (Y2H) system to detect direct protein–protein interactions in vivo. The full-length CDSs of PeTALE2, PeTALE5, and PeTALE7 were individually cloned into the pGADT7 activation domain vector at the NdeI restriction site. Similarly, the full-length CDSs of PeTALE4, PeTALE32, and PeTALE6 were cloned into the pGBKT7 DNA-binding domain vector at the NdeI site. The bait–prey pairs were co-transformed into the yeast strain AH109 using the Matchmaker™ GAL4 Two-Hybrid System (Takara Bio, USA, Inc., San Jose, CA, USA), alongside controls: pGADT7-T + pGBKT7-53 as the positive control and pGADT7-T + pGBKT7-lam as the negative control. After transformation, yeast cells were incubated at 28 °C for 3 days. To select for successful transformations, colonies were first plated onto selective medium (SD/-Leu-Trp) to ensure plasmid integration. Protein–protein interactions were then assessed by plating the transformants onto a more stringent selection medium (SD/-Leu-Trp-His-Ade) supplemented with 3-amino-1,2,4-triazole (3-AT) to inhibit non-specific background activation.
4.10. Statistical Analysis
The experimental data were analyzed using a one-way analysis of variance (ANOVA) with SPSS 24.0. All results are presented as the mean ± standard error. Significant differences (p < 0.05) between the control and treatment groups were further assessed using Student’s t-test.
5. Conclusions
This study provides a comprehensive analysis of the PeTALE gene family in Passiflora edulis, identifying 23 distinct PeTALE genes. Phylogenetic and syntenic analyses revealed that both tandem and segmental duplications contributed to the expansion and evolutionary diversification of the PeTALE gene family in passion fruit. Tissue- and stage-specific expression patterns suggest that PeTALE genes are crucial regulators of reproductive development, particularly in floral organ formation and in response to temperature stresses. Moreover, our findings highlight the critical role of PeTALE genes in coordinating floral organ development and modulating floral bud differentiation under varying temperature conditions. This work establishes a solid foundation for future research aimed at elucidating the molecular mechanisms by which PeTALE genes influence flower and fruit development, as well as their potential roles in stress adaptation.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Sablowski R. Plant stem cell niches: From signalling to execution Curr. Opin. Plant Biol.2011144910.1016/j.pbi.2010.08.00120739214 · doi ↗ · pubmed ↗
- 2Arnaud N. Pautot V. Ring the BELL and tie the KNOX: Roles for TAL Es in gynoeciurn development Front Plant Sci.201459310.3389/fpls.2014.0009324688486 PMC 3960571 · doi ↗ · pubmed ↗
- 3Hake S. Vollbrecht E. Freeling M. Cloning Knotted, the dominant morphological mutant in maize using Ds 2 as a transposon tag EMBO J.19898152210.1002/j.1460-2075.1989.tb 03343.x 16453866 PMC 400767 · doi ↗ · pubmed ↗
- 4Aichinger E. Kornet N. Friedrich T. Laux T. Plant stem cell niches Annu. Rev. Plant Biol.20126361563610.1146/annurev-arplant-042811-10555522404469 · doi ↗ · pubmed ↗
- 5Mukherjee K. Brocchieri L. Bürglin T.R. A Comprehensive Classification and Evolutionary Analysis of Plant Homeobox Genes Mol. Biol. Evol.2009262775279410.1093/molbev/msp 20119734295 PMC 2775110 · doi ↗ · pubmed ↗
- 6Hamant O. Pautot V. Plant development: A TALE story Curr. Biol.201033337138110.1016/j.crvi.2010.01.01520371112 · doi ↗ · pubmed ↗
- 7Hay A. Tsiantis M. KNOX genes: Versatile regulators of plant development and diversity Development 20101373153316510.1242/dev.03004920823061 · doi ↗ · pubmed ↗
- 8Bolduc N. Hake S. The maize transcription factor KNOTTED 1 directly regulates the gibberellin catabolism gene ga 2ox 1Plant Cell 2009211647165810.1105/tpc.109.06822119567707 PMC 2714931 · doi ↗ · pubmed ↗