The Role of Cardiac Magnetic Resonance Imaging in the Management of Hypertrophic Cardiomyopathy
Luca Pugliese, Alessandra Luciano, Marcello Chiocchi

TL;DR
This paper discusses how cardiac MRI helps diagnose and manage hypertrophic cardiomyopathy, a heart condition with varied symptoms and genetic causes.
Contribution
The paper highlights the growing role of contrast-enhanced cardiac MRI in diagnosing and managing hypertrophic cardiomyopathy.
Findings
CMR provides detailed cardiac morphology and function information in HCM patients.
CMR detects hypertrophy in areas not visible via echocardiogram and identifies myocardial fibrosis.
CMR aids in evaluating LV outflow tract obstruction and assessing LV function in advanced HCM.
Abstract
Hypertrophic cardiomyopathy (HCM) is the most common genetic cardiomyopathy, caused by either sarcomere protein or other gene mutations. It is a complex and highly heterogeneous disorder, with phenotypes ranging from asymptomatic to severe disease, characterized by asymmetric left ventricular (LV) hypertrophy unexplained by loading conditions, which is also associated with myocardial fiber disarray, and preserved or increased ejection fraction without LV dilation. Comprehensive personal and family history, physical examination, and ECG testing raise suspicion of HCM, and echocardiogram represents the first-line imaging modality for confirming a diagnosis. Moreover, contrast-enhanced cardiac magnetic resonance (CMR) imaging has increasingly emerged as a fundamental diagnostic and prognostic tool in HCM management. This article reviews the role of CMR in HCM identification and…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
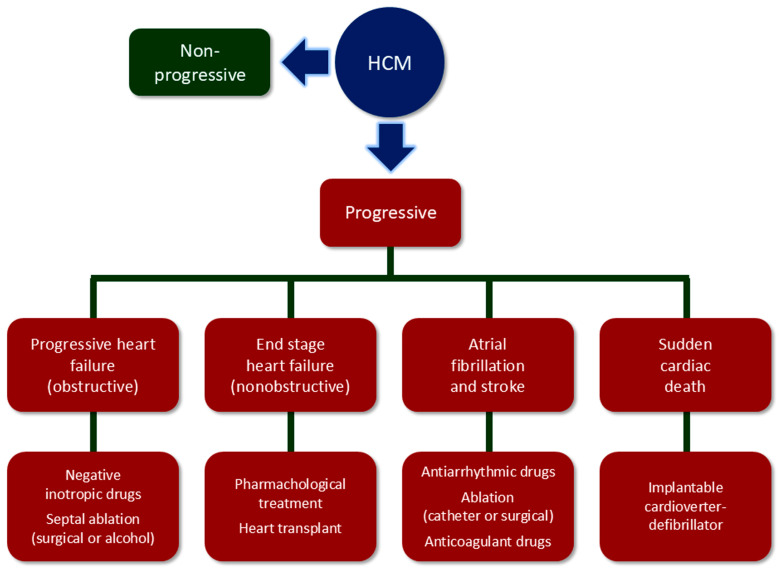 Figure 1
Figure 1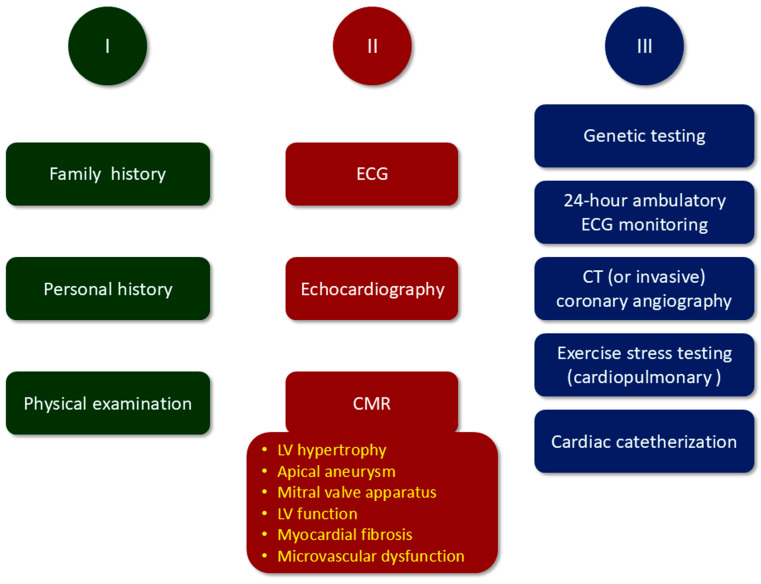 Figure 2
Figure 2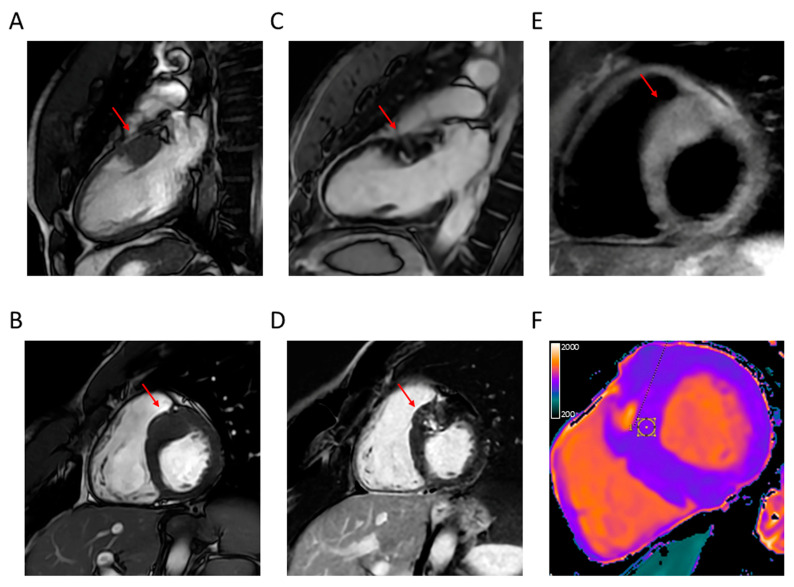 Figure 3
Figure 3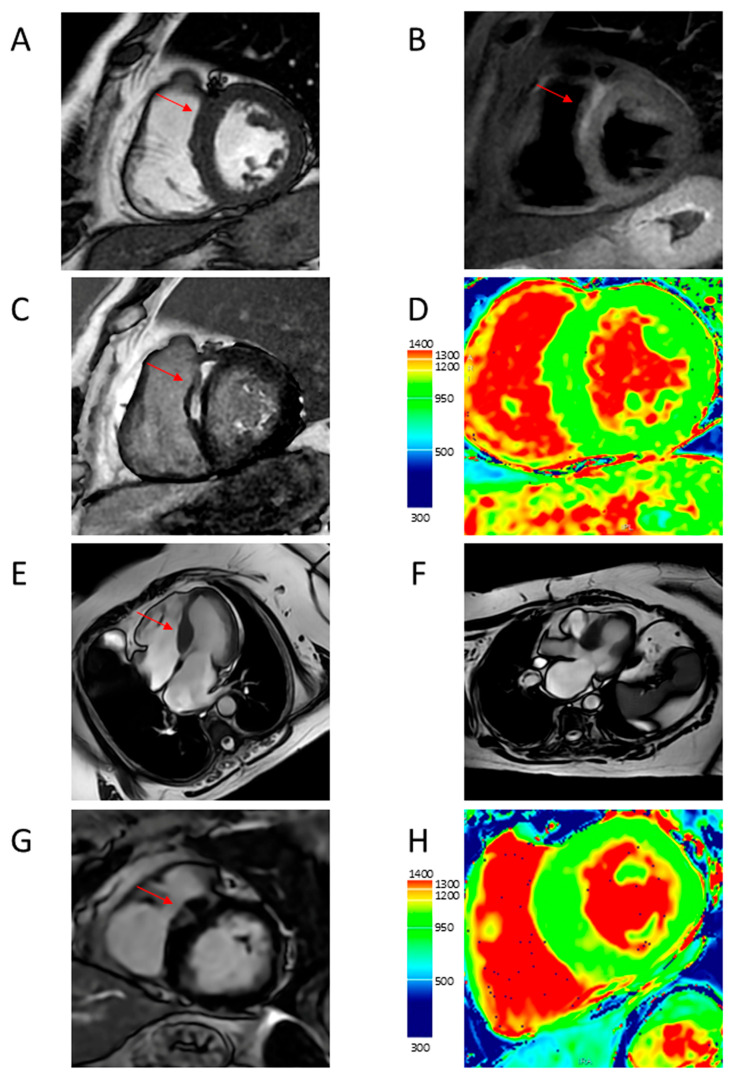 Figure 4
Figure 4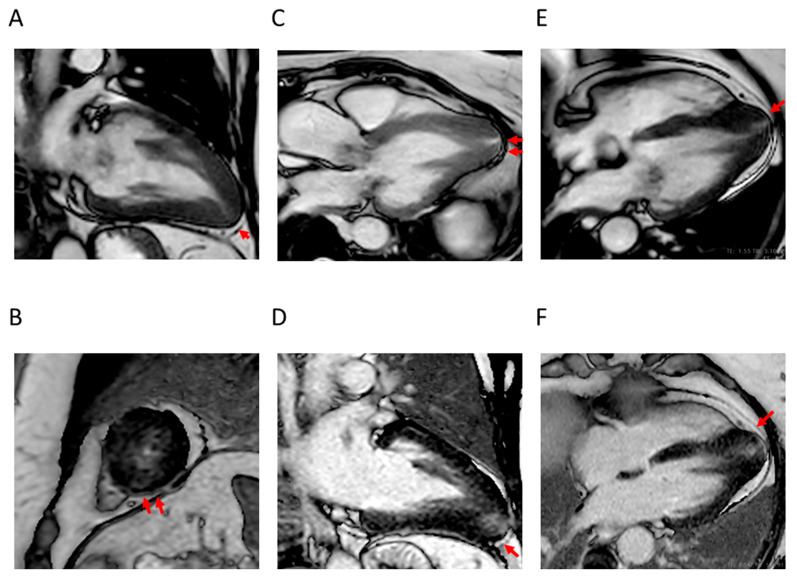 Figure 5
Figure 5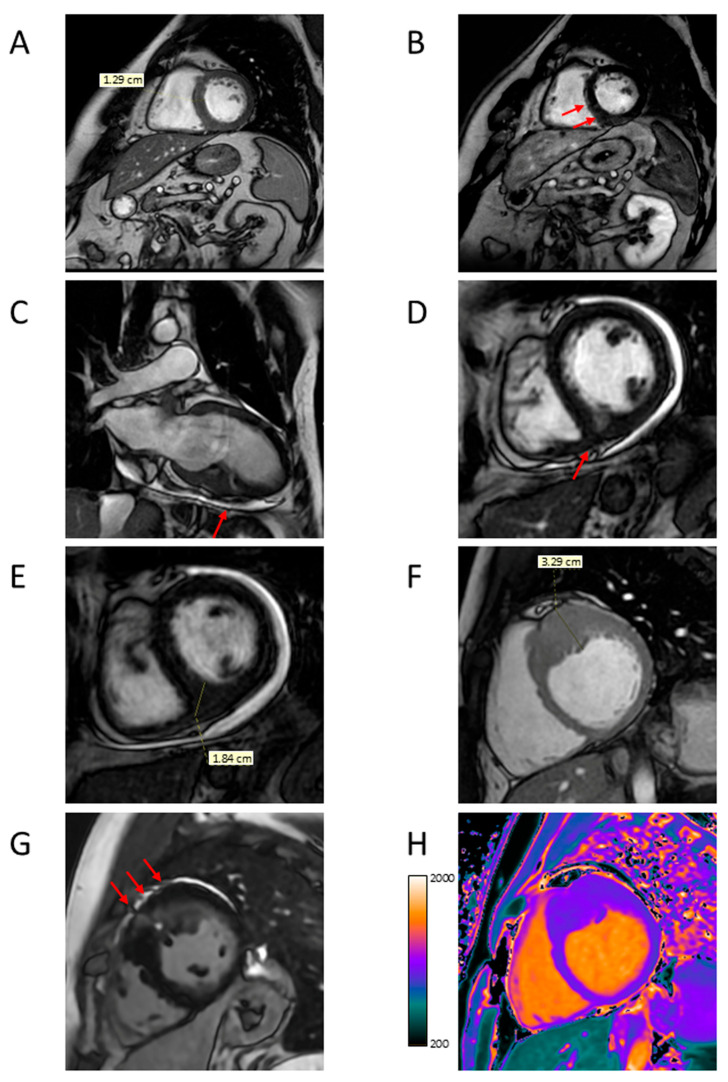 Figure 6
Figure 6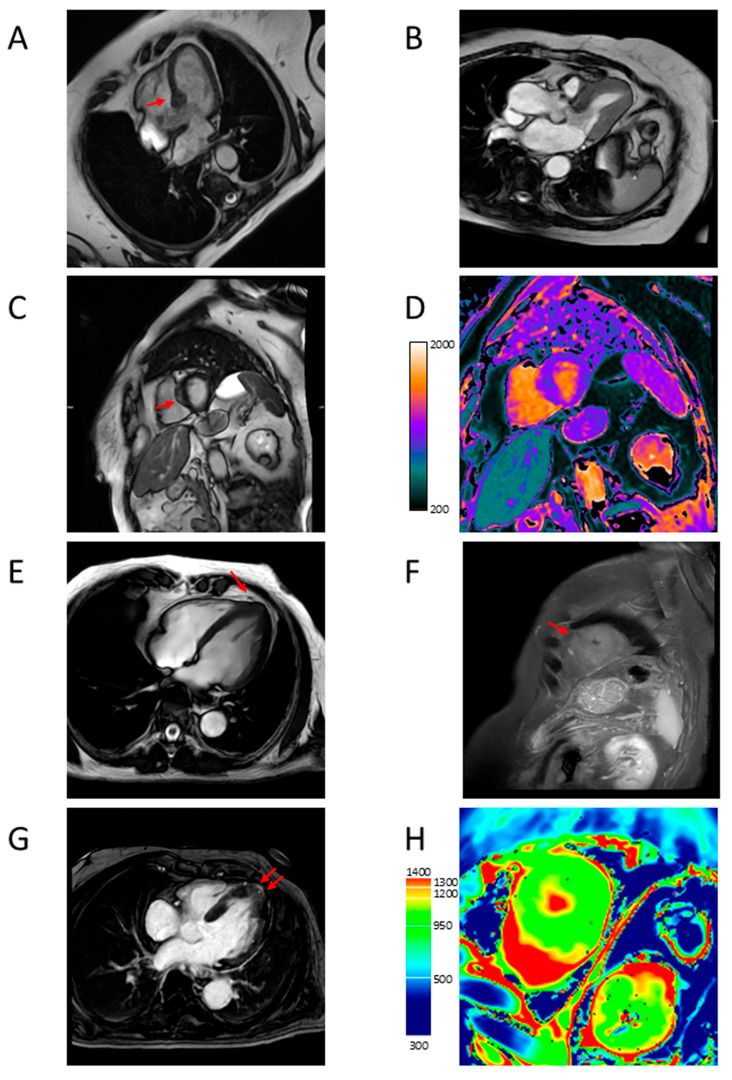 Figure 7
Figure 7Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsCardiomyopathy and Myosin Studies · Cardiovascular Function and Risk Factors · Cardiovascular Effects of Exercise
1. Introduction
Hypertrophic cardiomyopathy (HCM) is a genetic cardiomyopathy characterized by left ventricular (LV) hypertrophy. It has long been considered a rare and mostly serious disease with limited treatment options, predominantly affecting young males from the Western world [1]. However, in recent decades, the introduction of echocardiography has revealed that HCM is indeed a relatively common disorder with worldwide distribution and a wide age range, that affects both sexes and presents with a broad disease spectrum, from asymptomatic to severe [1]. Moreover, diagnostic and therapeutic innovations have become available, thus substantially decreasing morbidity and mortality from HCM and making it a treatable condition compatible with normal longevity [2]. This article will review the role of cardiac magnetic resonance (CMR) as a fundamental diagnostic and prognostic tool in HCM management, complementary to echocardiography.
2. Multifaceted Nature of Hypertrophic Cardiomyopathy (HCM)
The increasing recognition of the existence of multiple HCM phenotypes, from cases with subtle disease without significant disability to symptomatic forms eventually causing arrhythmic sudden cardiac death (SCD), atrial fibrillation (AF) and stroke, and heart failure, has made HCM the most common genetic cardiomyopathy with an estimated prevalence in the general population of 1:200 to 1:500 [3].
Though HCM is often inherited as a Mendelian autosomal dominant disease with variable penetrance and expression, variants in genes encoding sarcomere proteins involved in contractile function are found only in 30–40% of cases [4]. Several mutations of sarcomere genes have been identified so far. The two most common genes are those encoding for beta myosin heavy chain 7 (MYH7) and myosin-binding protein C3 (MYBPC3), while genes encoding for TNNI3, TNNT2, TPM1, MYL2, MYL3, and ACTC1 each account for ≤5% of patients. Although these eight variants show the strongest evidence for association with HCM [5], they do not always predict the clinical phenotype and prognosis, thus suggesting that the heterogeneity of HCM cannot be explained solely by sarcomere gene mutations [6]. The introduction of high-throughput sequencing techniques has also allowed the identification of non-sarcomere gene variants showing a moderate-to-strong association with HCM [4], and cases with autosomal recessive inheritance of these gene variants have been described, especially in populations with a higher degree of consanguinity [7].
The typical pathological feature of HCM is LV hypertrophy, which is predominantly asymmetric and involves the basal interventricular septum below the aortic valve, together with the LV free wall, and less frequently other LV segments and even the right ventricle (RV) [8]. Other features include elongation of the mitral valve leaflet(s) and abnormal insertion of the corresponding papillary muscle [9]. At the histological level, myocyte enlargement and disarray are often associated with various degrees of interstitial fibrosis [10].
Hypertrophic LV is characterized by increased wall thickness at the end-diastole (≥15 mm) at any site, but mostly at the basal anterior septal region (classical HCM pattern), with a ratio of septal to inferolateral wall thickness ≥ 1.3, associated with normal or small cavity and normal end-diastolic and reduced end-systolic volume. Less common variants include the reverse septal, neutral (uniform hypertrophic septum), midventricular, apical, and concentric hypertrophy [11,12]. The LV is typically hyperdynamic with high-normal or elevated ejection fraction [8]. Diastolic dysfunction occurs frequently in HCM as a consequence of interstitial fibrosis and the increased stiffness of the thickened LV wall [1,8]. Approximately 60–70% of individuals with HCM have LV outflow tract obstruction (LVOTO), with half of them exhibiting it at rest and the other half when provoked by stress [13,14]. It is dynamic in nature since it is favored by systolic anterior motion (SAM) and other abnormalities of the mitral valve that abut and obstruct the outflow tract at the subaortic level during systole [8]. Relatively mild segmental hypertrophy (13–15 mm) has been observed in some individuals, associated with normal LV mass and often with dynamic LVOTO [15]. The presence of LVOTO [16], as well as of concomitant severe coronary atherosclerosis [17], may exacerbate the mismatch between myocardial oxygen supply and demand, thus causing myocardial ischemia [18]. Decreased oxygen supply is due to microvascular dysfunction/injury resulting from structural (medial hypertrophy, intimal hyperplasia, and reduced capillary density) and functional abnormalities of the small vessels impairing coronary flow reserve, whereas increased oxygen demand is due to myocardial hypertrophy, with compression from diastolic dysfunction further reducing oxygen delivery [19].
Many HCM patients are asymptomatic or only mildly symptomatic and have a benign course with an extended lifespan. In these individuals, current guidelines for HCM management have even removed the universal restriction from vigorous physical activity or competitive sports, which can be considered following an annual comprehensive evaluation and discussion of the potential benefits and risks with an HCM expert [20].
However, other individuals experience unfavorable disease progression along one or more of four adverse pathways, although progression along two or three pathways occurs in only 10% of patients [21]. The clinical manifestations are the consequence of arrhythmias or systolic dysfunction and include SCD due to ventricular tachycardia or fibrillation, thromboembolic stroke due to AF, progressive heart failure in individuals with obstructive HCM, and advanced heart failure and end-stage in individuals with non-obstructive HCM [2,3,22]. It has been estimated that these four pathways account for 6%, 39%, 17%, and 4% of HCM cases, respectively, whereas the remaining present a benign or stable course [21,23] (Figure 1).
The prognosis of symptomatic HCM patients has significantly improved over the last decades due to the availability of effective preventive and therapeutic measures. Prophylactic use of intravenous implantable cardioverter-defibrillator (ICD), guided by risk stratification algorithms for candidate selection, is highly effective for preventing SCD and has been the main contributor to the observed ≥10-fold decrease in HMC-related mortality to 0.5% per year [2,3,24]. Invasive septal reduction treatments, including surgical septal myectomy and percutaneous alcohol septal ablation, are successful in relieving symptoms and blocking heart failure progression due to severe LVOTO, which is resistant to negative inotropic drugs (i.e., β-blockers, verapamil, disopyramide, and eventually cardiac myosin inhibitors) [25,26,27]. Atrial fibrillation may be anticipated by using a predictive model [28] and recurrent episodes may be reduced by ~50% with antiarrhythmic drugs and catheter or surgical ablation [29]. Moreover, prophylactic anticoagulant treatment is effective in reducing the risk of thromboembolic events [30]. Finally, even end-stage in patients with non-obstructive HCM, which is now responsible for most deaths, can be controlled using drugs, implantable defibrillators, cardiac resynchronization, and heart transplant [31].
Early detection and risk stratification, together with regular follow-up, are therefore essential for the timely adoption of appropriate measures for the successful prevention and treatment of HCM-related morbidity and mortality.
3. Diagnosis, Risk Stratification, and Follow-Up of HCM
Suspicion of HCM may arise from the presence of a family history of HCM or SDC, typical signs and symptoms, or abnormal results of a routine electrocardiogram (ECG) or echocardiography performed for other indications. According to current guidelines for HCM management [20], initial evaluation should indeed include a comprehensive personal and family history and physical examination, followed by a 12-lead ECG and cardiac imaging (Figure 2).
A detailed family history should include at least three generations. In addition, genetic testing is important for facilitating the identification of family members at risk of developing HCM and for diagnosing patients with an atypical clinical presentation [32,33].
When present, symptoms include exertional dyspnea, exercise intolerance, orthopnea, palpitations, presyncope, and syncope, which may be exertional or immediately post-exertional in the case of significant LVOTO, and chest pain [8].
Physical examination should be performed at rest and after maneuvers such as Valsalva and squat-to-stand, in order to detect signs of LVOTO. Signs include a harsh crescendo–decrescendo systolic murmur, prominent apical point of maximal impulse, abnormal carotid pulse, and a fourth heart sound.
The ECG is often abnormal, even in patients with no or only mild LVOTO. Abnormalities comprise voltage changes suggestive of LV hypertrophy and eventually evidence of left atrial enlargement, ST segment and T wave abnormalities, and deep Q waves [8].
Two-dimension (2D) transthoracic echocardiography is the first-line imaging modality for confirming HCM diagnosis and identifying the disease phenotype, thus allowing risk stratification [34,35]. Indeed, it allows us to determine the presence, extent, and pattern of LV hypertrophy, the hallmark of HCM, and also to detect apical aneurysms, assess the presence and severity of LVOTO, and evaluate LV systolic and diastolic and mitral valve function. Provocative maneuvers, such as Valsalva or squat-to-stand (stress echocardiography), should be performed if the resting gradient is <50 mm Hg, to unmask the presence of LVOTO, as it is not significant at rest in half of patients presenting with this complication [36]. Screening with ECG and 2D echocardiography is indicated also in asymptomatic family members of HCM patients [20].
By providing complementary information and as an alternative to echocardiography for patients with inconclusive echocardiogram, CMR imaging has increasingly emerged as a fundamental diagnostic and prognostic tool in HCM management [21].
Other diagnostic procedures are also needed for further characterization of HCM phenotype (Figure 2). If echocardiography is not diagnostic and CMR imaging is unavailable, cardiac computed tomography (CT) may be considered [20], whereas CT (or invasive) coronary angiography is recommended in patients with myocardial ischemia and those at risk for coronary atherosclerosis before surgical septal myectomy [20]. A 24-h ambulatory ECG monitoring is recommended in HCM patients with arrhythmias and at risk for SCD, whereas exercise stress testing (or cardiopulmonary exercise stress testing) is indicated to determine functional capacity [20]. Finally, if the presence or severity of LVOTO is not adequately assessed or the hemodynamic profile is not sufficiently characterized by noninvasive imaging studies, invasive hemodynamic assessment with cardiac catheterization is recommended [20].
Medical history, physical examination, ECG, 2D transthoracic echocardiography, and CMR should be repeated at variable intervals, depending on the presence and severity of symptoms and signs, to monitor the course of the disease [20].
4. Cardiac Magnetic Resonance (CMR) Features of HCM
High-field scanners, such as 1.5 T or 3 T, are required for performing CMR. End-expiration breath-hold images are acquired with ECG triggering, and heart long and short axes are localized with scout sequences. Short-axis slices of the LV from the mitral valve to the apex and two-, three- and four-chamber long-axis views of the heart are then acquired and sequences are repeated after the administration of a gadolinium-based contrast agent [37]. The CMR acquisition protocol and sequences, according to the Society for Cardiovascular Magnetic Resonance guidelines [38], are reported in Table 1. Application of a deep learning algorithm may improve the performance of CMR for HCM diagnosis [39], especially if combined with radiomics [40].
By the use of several techniques, CMR has the advantages over echocardiography of a high spatial and temporal resolution, the provision of tomographic images of the heart without a limited view, and the unique ability to identify myocardial fibrosis [41]. Cine-balanced steady-state free precession (SSFP) imaging allows an accurate evaluation of cardiac morphology and function [42]. Late gadolinium enhancement (LGE) CMR imaging using a segmented inversion recovery pulse sequence is able to differentiate between normal and infarcted or fibrotic myocardium [43]. T1 and T2 mapping CMR imaging with end-diastolic Modified Look-Locker Inversion Recovery (MOLLI) sequences are also able to detect areas of myocardial injury without injection of gadolinium-based contrast agents through the assessment of the total extent of expanded extracellular space. Native T1 values are increased in areas of myocardial scarring and interstitial fibrosis and T1 mapping before and after contrast medium injection allows calculation of the extracellular volume fraction [43]. T2-weighted imaging with double inversion recovery black blood preparation can identify areas of myocardial edema or inflammation and T2 mapping can confirm myocardial hyperintensity [44]. Abnormal T1 and T2 values may be observed in areas with or without LGE [41]. Less used techniques include cine phase contrast, perfusion, and tagging CMR imaging, which may serve for the assessment of blood flow and evaluation of myocardial wall motion and strain [41].
These characteristics make CRM superior to echocardiography for the identification of LV hypertrophy, apical aneurysm, and structural abnormalities of the mitral valve and subvalvular apparatus contributing to LVOTO and the evaluation of LV function, in addition to allowing the assessment of myocardial fibrosis and microvascular dysfunction (Figure 3, Figure 4, Figure 5, Figure 6 and Figure 7).
4.1. LV Hypertrophy
Due to its ability to provide a sharp contrast between the blood pool and myocardium and to distinguish LV from RV and other structures, cine-balanced SSFP CMR imaging with retrospective gating allows an accurate measurement of LV wall thickness, together with chamber size and mass [45].
In addition, because of its no-limited view, it allows the detection of hypertrophy in areas blind to echocardiogram, such as the apex, midventricular region, posterior septum, anterolateral free wall, and RV, as well as asymmetric hypertrophy with a spiral configuration and mass-like focal hypertrophy [46,47,48,49].
Finally, CMR is capable of identifying subtle morphologic features in the absence of LV hypertrophy, such as narrow blood-filled myocardial crypts, apical pouching or thinning, elongated mitral leaflets, and expanded extracellular space, which are typically seen in genotype positive/phenotype negative individuals [50,51,52,53].
4.2. Apical Aneurysm
Apical aneurysm has been related to myocardial ischemia and is associated with either apical hypertrophy, with the typical spade-like configuration of the LV cavity [54], or midventricular hypertrophy [55].
The presence of an LV apical aneurysm often goes unrecognized with echocardiography, although it may be detected with contrast echocardiography [56]. However, CMR is the gold standard for detecting apical aneurysms, which appear as a thin-walled dyskinetic or akinetic segment with a transmural scar and, eventually, the presence of a thrombus in the aneurysm cavity [57], which was shown to be associated with thromboembolic stroke [54,56].
4.3. LVOTO
Echocardiography is highly useful in the characterization of dynamic LVOTO by allowing estimation of the peak LV outflow tract gradient and assessment of the role of the mitral valve [20]. However, CMR also provides important insights into LVOTO evaluation [57] by allowing a precise assessment of the LV outflow tract and the abnormalities of the mitral valve apparatus contributing to it [58,59,60].
Cine phase contrast CMR imaging is a valuable technique for the assessment of LVOT severity by allowing the quantification of the blood flow through the LV outflow tract and the estimation of the gradient [61], although this is a cumbersome and time-consuming procedure, and hence, is not often used in the clinical setting [41].
Moreover, CMR is able to detect abnormalities of the mitral valve apparatus that are frequently missed by echocardiography. These abnormalities include elongated mitral valve leaflets and muscle bundles, hypertrophic papillary muscles, an increased number of papillary muscles, and anomalous insertion of the anterior papillary muscle to the mitral valve [52,62,63]. A common alteration in patients with HCM is the anterior and apical displacement of the base of the papillary muscle, usually the antero-lateral one, which causes leaflet slack resulting in SAM of the mitral valve and consequent LVOTO independent of apical hypertrophy [64]. Therefore, it is important to differentiate apical displacement of the papillary muscle from apical hypertrophy, with CRM being the gold standard, although contrast echocardiography can be also useful [65]. However, it has been recently shown that apical displacement of the papillary muscle may precede apical hypertrophy, suggesting a pathogenic link between these two conditions [66].
4.4. LV Function
Nowadays, CMR is the gold standard for the assessment of LV function due to its superior reproducibility for volumetric assessment when compared with echocardiography, which is limited by 2D geometric assumptions and the necessity of an adequate acoustic window [67,68].
Assessment of LV function is particularly important for risk stratification and disease monitoring since HCM is usually characterized by normal or supernormal LV ejection fraction, which, however, may decline below 50% in approximately 5–10% of patients who progress to the so-called end-stage (or burn-out) HCM, characterized by LV wall thinning and cavity dilation [69].
4.5. Myocardial Fibrosis
The unique ability of CMR to provide an in vivo characterization of myocardial tissue with the detection of interstitial fibrosis by the use of LGE and T1 mapping supports a major diagnostic and prognostic role for this imaging procedure in HCM.
At present, LGE is the gold standard for fibrosis detection [57] and is currently used for risk stratification of HCM [70]. However, while the presence of LGE has been associated with adverse outcomes in HCM [71,72,73], there is no consensus on which of the various methods for LGE quantification should be used. These methods, which include visual assessment, full width at half maximum, or signal intensity cut-off values of 2 to 7 standard deviations exceeding the mean value of the non-injured myocardium [43], have indeed shown to provide different LGE measures [74,75,76], and hence, different prognostic estimates, although a meta-analysis showed comparable accuracy in predicting SCD [77]. Around half of individuals with HCM have LGE, which usually appears as a patchy mid-myocardial area within the segment(s) of maximal LV hypertrophy [78,79]. However, LGE can identify focal, but not diffuse fibrosis, which can be detected only by T1 mapping [43]. This represents a promising CMR technique [80,81] that may prove to be superior to LGE, although its clinical relevance has not yet been established [57]. Moreover, it allows estimation of the extracellular volume (ECV) from post and pre-contrast T1 values of blood and myocardium and the level of hematocrit [82].
4.6. Microvascular Dysfunction
The use of CMR perfusion imaging allows the presence and extent of microvascular dysfunction to be identified [19].
Adenosine-stress perfusion defects on CMR were indeed shown in more than 40% of HCM patients [83]. Abnormal perfusion was associated with hypertrophy and LGE, although it was detected even in non-hypertrophic and non-LGE segments [84,85,86].
5. Role of CMR Imaging in HCM Management
Due to its ability to assess the main typical features of HCM, CMR offers a valuable contribution to the diagnosis and monitoring of the disease as well as to the differentiation of CMR from phenotypic mimics [87,88]. Moreover, it is useful for the characterization of HCM phenotypes and risk stratification, which is essential for predicting outcomes and making therapeutic decisions, including the use of ICD or septal reduction treatments.
5.1. Diagnosis and Differentiation from Phenotypic Mimics
The importance of CMR in HCM diagnosis relies on its ability to identify LV hypertrophy, the hallmark of the disease, in a more reliable and accurate manner than echocardiography. While echocardiography may either underestimate or overestimate LV wall thickness, CMR provides precise measurements and is also able to detect hypertrophy in areas blind to echocardiogram or even pre-hypertrophic lesions [45,46].
For the same reasons; CMR has been increasingly utilized in the screening of first-degree relatives of patients with HCM. Moreover; CMR was found to differentiate a positive from a negative genotype of HCM [89]. Screening should be performed with ECG and echocardiography every 1–3 years during childhood and adolescence and every 3–5 years in adult age; but CMR has also a role due to its ability to identify subtle myocardial abnormalities and LGE in the absence or prior to the presence of LV hypertrophy [20]
The suspicion of HCM is based on the presence of LV hypertrophy disproportionate to loading stimuli, at variance with hypertensive cardiomyopathy and aortic stenosis, but is similar to several conditions that mimic HCM, such as the athlete’s heart and infiltrative cardiomyopathies [90,91,92]. Although distinction from these conditions is based on a comprehensive and multiparametric workup, including the search for extracardiac manifestations, specific laboratory and genetic testing, and eventually myocardial biopsy, CMR provides important clues to differential diagnosis [20], which may also take advantage of the analysis of pressure-strain loop-derived myocardial work by speckle tracking echocardiography [93]. This is because of its ability to accurately detect the presence and pattern of LV hypertrophy and quantify the extent of LV wall thickening [45,46], combined with the identification of myocardial interstitial fibrosis [43]. The asymmetric distribution of LV hypertrophy is the main criterion for distinguishing HCM from most of its phenotypic mimics or phenocopies, but patients with HCM may also have a symmetric pattern [41].
In the athlete’s heart, symmetrical LV hypertrophy can be either eccentric or concentric depending on the type of training (endurance versus strength). This condition can be differentiated from HCM because it typically regresses with deconditioning (>2 mm reduction of LV wall thickness after a detraining period) [94] and is characterized by a diastolic wall thickness to left ventricular end-diastolic volume ratio <0.15 mm/m^2^/mL [95]. Moreover, the presence of areas of myocardial scarring/fibrosis at contrast-enhanced CMR with LGE is not consistent with the athlete’s heart, although a mesocardiac LGE stria has been rarely found at the anterior or posterior interventricular junctions and the insertion point of the trabeculae on the ventricular wall [96]. Finally, T1 mapping and ECV values are decreased instead of increased in the athlete’s heart, due to the predominant increase in the cellular over the extracellular myocardial component.
In hypertensive cardiomyopathy, LV hypertrophy is typically moderate, symmetric, and concentric, may regress with aggressive anti-hypertensive treatment, and is usually not associated with LVOTO due to SAM of the mitral valve, although it may be occasionally present [97,98]. In addition, myocardial crypts are not uncommon [99], whereas LGE is absent [100] or shows a linear or patchy pattern at the septal or inferior wall [41] and native T1 and T2 mapping and ECV are either normal or slightly increased [98,101]. Hypertrophy is symmetric also in aortic stenosis, which can be easily identified by CMR [90].
Infiltrative cardiomyopathies include amyloidosis, sarcoidosis, and glycogen/lysosomal storage diseases, such as Fabry–Anderson disease. In amyloidosis, LV hypertrophy is asymmetric in transthyretin amyloidosis and symmetric in immunoglobulin light-chain amyloidosis. Moreover, a right atrial free wall thickness >6 mm is considered to be a specific feature of cardiac amyloid [102]. Diffuse and subendocardial LGE characterizes the early stages, whereas transmural LGE is observed in the advanced stages of the disease [103]. Native T1 mapping and ECV are typically increased and T2 mapping values are also elevated, reflecting myocardial edema [104]. In sarcoidosis, typical findings differ in the acute (inflammatory) and chronic (fibrotic) phases of the disease. In the acute phase, there is LV hypertrophy with signal hyperintensity on T2-weighted imaging and wall motion abnormalities not corresponding to coronary distribution, whereas in the chronic phase, wall thinning or aneurysm are associated with signal hypointensity on T2-weighted imaging and the presence of LGE [105,106]. The distribution of LGE is typically at the level of the septal and lateral wall of basal segments [106], with a pattern predominantly mid-myocardial or subepicardial (non-ischemic) [107]. In Fabry–Anderson disease, LV hypertrophy is diffuse and asymmetrical and is associated with linear or patchy LGE at the basal-inferolateral mid-wall, decreased native T1 mapping values, at variance with other cardiomyopathies, and increased T2 mapping and ECV values [108,109,110].
5.2. Characterization of HCM Phenotypes and Risk Stratification
According to current guidelines [20], patients with HCM, adults, children, or adolescents, should undergo comprehensive, systematic noninvasive SCD risk assessment at initial evaluation and every 1 to 2 years thereafter in order to establish the need for ICD. This evaluation should include a personal history of cardiac arrest or sustained ventricular arrhythmias; personal history of syncope suspected by clinical history to be arrhythmic; family history in a close relative of premature HCM-related SCD, cardiac arrest, or sustained ventricular arrhythmias; maximal LV wall thickness, LV ejection fraction, LV apical aneurysm; non-sustained ventricular arrhythmias on continuous ambulatory ECG monitoring; left atrial diameter; and maximal LV outflow tract gradient.
In those patients with HCM who are not otherwise identified as high risk for SCD or in whom a decision to proceed with ICD placement remains uncertain, CMR imaging should be used for a more precise assessment of maximum LV wall thickness, LV ejection fraction, LV apical aneurysm, and possibly LV outflow tract gradient and for determining the extent of myocardial fibrosis with LGE, which is a strong predictor of ventricular arrhythmias [111]. This recommendation is supported by the demonstration of the prognostic significance of CMR assessment of these parameters [112,113,114,115,116,117,118]. The cutoffs are ≥30 mm maximum LV wall thickness in any segment within the chamber (≥28 mm in individual patients, not established for pediatric patients), <50% for LV ejection fraction, ≥50 mm Hg for LV outflow tract gradient, and ≥15% of LV mass for LGE (not established for pediatric patients), whereas the presence of apical aneurysm is relevant independent of size [20].
This information may be used to calculate the 5-year risk of SCD using an SCD risk algorithm such as that of the American College of Cardiologists American Heart Association, which includes age, family history of SCD, maximum LV wall thickness, left atrial diameter, maximal LV outflow tract gradient, non-sustained ventricular tachycardia, unexplained syncope, apical aneurysm, and extensive LGE [119].
By providing important information on LV outflow tract gradient and mitral valve apparatus as well as on LV systolic function, CMR is useful not only for calculating SCD risk but also for assessing the progression of heart failure due to LVOTO (obstructive) and LV systolic dysfunction (non-obstructive). As mentioned above, CMR plays a critical role in identifying abnormalities of the mitral valve apparatus undetected at echocardiography. The identification of these abnormalities is crucial for choosing septal reduction treatments, since they are corrected by surgical septal myectomy but not by percutaneous alcohol septal ablation [58]. In addition, according to current guidelines [120], CMR represents an important imaging tool in the management of heart failure in patients with HCM. In these individuals, detecting an initial decrease in LV ejection fraction may indeed allow prompt recognition and treatment of this condition.
6. Conclusions
Contrast-enhanced CMR has increasingly emerged as an indispensable diagnostic and prognostic tool in patients with suspected or known HCM by providing important insights into the phenotype characterization and risk stratification of these individuals, thus guiding the choice of preventive and therapeutic measures.
In particular, it allows more accurate detection and quantification of cardiac morphological and functional abnormalities than 2D-echocardiography, including LV hypertrophy, apical aneurysm, structural abnormalities of the mitral valve and subvalvular apparatus, and LV dysfunction. In addition, it provides information on the presence and extent of myocardial fibrosis, which represents a powerful predictor of adverse outcomes in HCM due to its major role in the pathogenesis of arrhythmias and systolic dysfunction.
Therefore, in order to better manage HCM and its multifaceted manifestations, the use of CMR in clinical practice should be further expanded, and the ability of CMR measures to predict disease progression and adverse outcomes should be evaluated in more depth.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Maron B.J. Rowin E.J. Maron M.S. Hypertrophic Cardiomyopathy: New Concepts and Therapies Annu. Rev. Med.20227336337510.1146/annurev-med-042220-02153935084989 · doi ↗ · pubmed ↗
- 2Maron B.J. Rowin E.J. Casey S.A. Maron M.S. How Hypertrophic Cardiomyopathy Became a Contemporary Treatable Genetic Disease with Low Mortality: Shaped by 50 Years of Clinical Research and Practice JAMA Cardiol.201619810510.1001/jamacardio.2015.035427437663 · doi ↗ · pubmed ↗
- 3Maron B.J. Clinical Course and Management of Hypertrophic Cardiomyopathy N. Engl. J. Med.201837965566810.1056/NEJ Mra 171057530110588 · doi ↗ · pubmed ↗
- 4Lopes L.R. Ho C.Y. Elliott P.M. Genetics of hypertrophic cardiomyopathy: Established and emerging implications for clinical practice Eur. Heart J.2024452727273410.1093/eurheartj/ehae 42138984491 PMC 11313585 · doi ↗ · pubmed ↗
- 5Ingles J. Goldstein J. Thaxton C. Caleshu C. Corty E.W. Crowley S.B. Dougherty K. Harrison S.M. Mc Glaughon J. Milko L.V. Evaluating the Clinical Validity of Hypertrophic Cardiomyopathy Genes Circ. Genom. Precis. Med.201912 e 00246010.1161/CIRCGEN.119.00246030681346 PMC 6410971 · doi ↗ · pubmed ↗
- 6Maron B.J. Maron M.S. Maron B.A. Loscalzo J. Moving Beyond the Sarcomere to Explain Heterogeneity in Hypertrophic Cardiomyopathy: JACC Review Topic of the Week J. Am. Coll. Cardiol.2019731978198610.1016/j.jacc.2019.01.06131000001 PMC 6550351 · doi ↗ · pubmed ↗
- 7Allouba M. Walsh R. Afify A. Hosny M. Halawa S. Galal A. Fathy M. Theotokis P.I. Boraey A. Ellithy A. Ethnicity, consanguinity, and genetic architecture of hypertrophic cardiomyopathy Eur. Heart J.2023445146515810.1093/eurheartj/ehad 37237431535 PMC 10733735 · doi ↗ · pubmed ↗
- 8Marian A.J. Braunwald E. Hypertrophic Cardiomyopathy: Genetics; Pathogenesis; Clinical Manifestations; Diagnosis; and Therapy Circ. Res.201712174977010.1161/CIRCRESAHA.117.31105928912181 PMC 5654557 · doi ↗ · pubmed ↗