The Expression of a Germline Fusion Gene Involving a Protein-Coding and a Long Non-Coding RNA Gene Results in Severe Brain Malformations
Lukas Kaufmann, Christine Beichler, Jasmin Blatterer, Ingrid Janisch, Bence Csapó, Elisabeth Schreiner, Sarah Verheyen, Jochen B. Geigl, Christian Windpassinger

TL;DR
A rare genetic fusion involving a protein-coding gene and a long non-coding RNA gene causes severe brain malformations in a fetus.
Contribution
First report of a germline fusion between a protein-coding and lncRNA gene resulting in a neomorphic protein linked to severe disease.
Findings
Germline fusion of MN1 and CPMER genes was identified as the cause of severe cerebral abnormalities.
Expression of C-terminally truncated MN1 proteins was observed, similar to a known gain-of-function syndrome.
The study highlights the potential role of lncRNA-involved gene fusions in disease-related protein expression.
Abstract
In the present study, an exceptional germline gene fusion involving the protein-coding MN1 gene and the long non-coding RNA (lncRNA) gene CPMER was detected as the genetic cause of severe cerebral abnormalities with unfavorable prognosis in a male fetus at 14 weeks of gestation. Quantitative and qualitative RNA analyses indicate the expression of C-terminally truncated MN1 proteins. MN1 proteins lacking the C-terminal amino acids have been previously described to cause an ultra-rare syndrome with brain malformations due to a gain-of-function effect. To the best of our knowledge, this is the first study reporting a germline gene fusion of a protein-coding gene and an lncRNA gene linked to a functional, but neomorphic, protein associated with severe phenotypic abnormalities. The results of our study are not only relevant for the genotype–phenotype correlation of MN1 but should especially…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
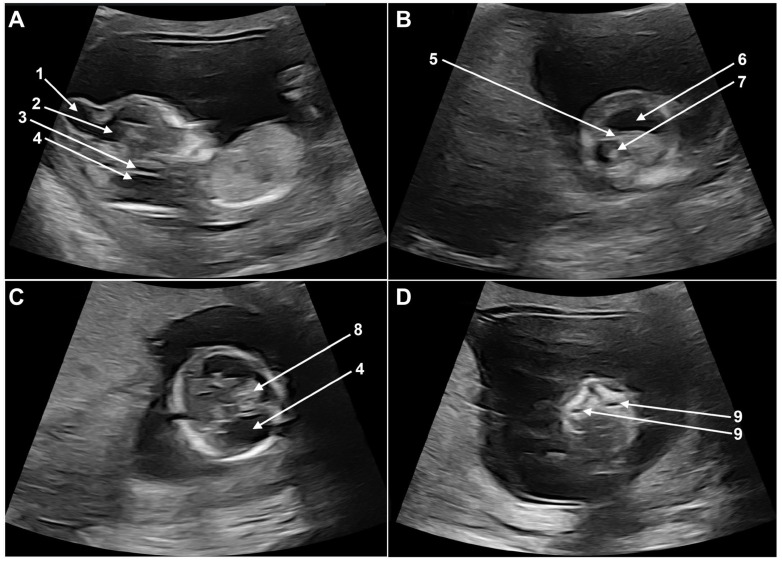 Figure 1
Figure 1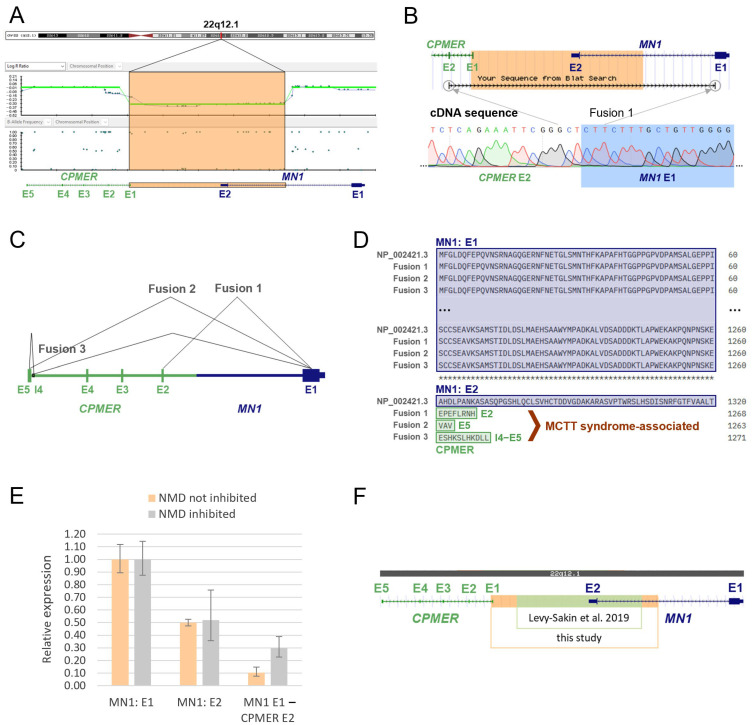 Figure 2
Figure 2Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsCancer-related molecular mechanisms research · RNA modifications and cancer · RNA Research and Splicing
1. Introduction
Gene fusions can be the result of chromosomal rearrangements such as deletions, translocations, tandem duplications, or inversions, as well as the splicing of a chimeric pre-mRNA [1]. Fusions involving long non-coding RNA (lncRNA) genes are not yet well understood but are thought to be highly relevant in tumorigenesis. Recently, it has been discovered that fusions of protein-coding genes (PCGs) with lncRNA genes can even be translated into functional proteins. However, this phenomenon has only been described in somatic cancer cells so far [2,3].
MN1 C-terminal truncation (MCTT) syndrome is a very rare disease with fewer than 30 cases described worldwide, characterized by craniofacial defects, expressive language delay, impaired intellectual development, dysmorphic ears, and distinct structural brain abnormalities [4]. The syndrome is caused by truncating variants in MN1 through a gain-of-function effect, mediated by the expression of MN1 proteins lacking the C-terminal amino acids encoded by exon 2 [5,6]. The MN1 proto-oncogene, transcriptional regulator (MN1) is a protein-coding gene located in the chromosomal region 22q12.1. MN1 comprises only two exons, with the majority (95%) of the 1320 amino acid long MN1 protein being encoded by exon 1 [7]. To date, exclusively C-terminal nonsense and frameshift variants escaping nonsense-mediated mRNA decay have been reported in association with MCTT syndrome [5,6].
In the present study, an exceptional germline gene fusion involving MN1 and an lncRNA was detected in a male fetus displaying severe cerebral abnormalities.
2. Materials and Methods
2.1. Ethics Statement
This study was approved by the ethics committee of the Medical University of Graz, Austria (approval number: 35-476 ex 22/23). Written informed consent was obtained from the mother of the fetus.
2.2. Clinical Evaluation and Diagnosis
The initial referral of the pregnant woman and the findings of the sonographic examination of the fetus were provided by experts of the Department of Obstetrics and Gynaecology at the University Hospital of Graz, Austria.
2.3. DNA and RNA Isolation
DNA isolation was carried out with phenol–chloroform from chorionic villus using a standard protocol. RNA was isolated from chorionic villus culture using TRIzol reagent (Invitrogen™/Thermo Fisher Scientific, Waltham, MA, USA). Some of the cultures were pre-treated with puromycin solution (catalog no. P8833, Sigma-Aldrich, Burlington, MA, USA) to inhibit nonsense-mediated mRNA decay (NMD), according to the standard operating procedure (SOP) “RNA Analyse nach KurzzeitKul_Trizol_isol” of our institute.
2.4. SNP Array
Copy number variation analysis was performed using the Infinium CytoSNP-850K (Illumina, San Diego, CA, USA) in combination with the BlueFuse™ Multi Analysis Software v4.5 (Illumina). The obtained data were also analyzed using the UCSC Genome Browser (www.genome.ucsc.edu, accessed on 24 November 2024) [8] and the DECIPHER database [9,10].
2.5. Quantitative Real-Time PCR
Quantitative real-time PCR (qRT-PCR) analysis from DNA was performed using a 7500 Fast Real-Time PCR System (Applied Biosystems™/Thermo Fisher Scientific, Waltham, MA, USA). With qRT-PCR from DNA, confined placental mosaicism was excluded, the size of the deletion detected by SNP array was confirmed, and segregation analysis on the parents of the fetus was performed. The region-specific primer sets used are listed in Supplementary Table S1.
For quantitative RNA analysis, qRT-PCR from cDNA was performed. RNA was quantified using the Qubit™ RNA Broad Range Assay Kit (catalog no. Q10210, Invitrogen™/Thermo Fisher Scientific): RNA_NMD not inhibited = 145 ng/μL and RNA_NMD inhibited = 179 ng/μL. The integrity of the RNA was then checked using the Bioanalyzer RNA 6000 pico assay (catalog no. 5067-1513, Agilent, Santa Clara, CA, USA) according to the manufacturer’s protocol, and the 18S and 28S RNA peaks were visible. Subsequently, cDNA synthesis was conducted by reverse transcription with the Omniscript RT Kit (catalog no. 205113, Qiagen, Hilden, Germany) according to the manufacturer’s protocol, using 870 ng of RNA_NMD not inhibited and 895 ng of RNA_NMD inhibited. The qRT-PCR analysis of the cDNAs of RNA_NMD not inhibited, RNA_NMD inhibited, and the Human Reference RNA (catalog no. 750500, Agilent) was performed with the StepOnePlus™ Real-Time PCR System (catalog no. 4376600, Applied Biosystems™/Thermo Fisher Scientific), using a standard protocol with 40 cycles, as well as melting curve analysis. The amplicon of exon 1 of MN1 was used as a comparative control for the quantitation of comparative Ct (delta–delta Ct values) analysis. The primer sets used for quantitative RNA (cDNA) analysis are listed in Supplementary Table S2.
2.6. Sanger Sequencing of cDNA
QuantiTect Reverse Transcription Kit (catalog no. 205311, Qiagen) was used for cDNA synthesis according to the manufacturer’s protocol. Subsequent amplification of the cDNA was performed using AmpliTaq GoldTM 360 Master Mix (catalog no. 4398881, Applied Biosystems™/Thermo Fisher Scientific) according to the SOP “AmpliTaq Gold 360 MasterMix PCR” of our institute. The primer sets listed in Supplementary Table S3 were used for Sanger sequencing of the cDNA samples “RNA_NMD not inhibited” (without prior puromycin treatment), “RNA_NMD inhibited” (with prior puromycin treatment), and the Human Reference RNA (catalog no. 750500, Agilent), which was utilized as control sample. The primer sets “sp_MN1_E1_v2_f” and “sp_MN1_E1_r” were used as a positive control, as exon 1 of the MN1 gene is not affected by the deletion. The wild-type sequence of this region in exon 1 was confirmed in both RNA_NMD not inhibited and RNA_NMD inhibited, as well as in the control sample. The remaining primer sets were used to detect possible fusion genes and their expression. In addition, information on splicing was obtained. For protein sequence alignment, the analysis tool Clustal Omega v2.1 [11] was used.
2.7. Trio-Whole-Exome Sequencing
Trio-whole-exome sequencing (Trio-WES) was performed in addition to exclude the possibility of other pathogenic variants in protein-coding genes that could be the cause of the fetal phenotype abnormalities. Trio-WES involves the sequencing of all protein-coding regions of the genome as well as the mitochondrial DNA, with subsequent phenotype-based screening and comparison with the parents’ data. Variant prioritization was based on the HPO terms: intrauterine growth retardation (HP:0001511), decreased thalamic volume (HP:0012695), encephalocele (HP:0002084), intracranial cystic lesion (HP:0010576), aplasia/hypoplasia of the brainstem (HP:0007362) and ventriculomegaly (HP:0002119). The specific regions were enriched using Twist Human Core Exome and Twist Mitochondrial Panel (TWIST Bioscience, South San Francisco, CA, USA). Sequencing was performed on the NextSeq 550 Sequencing System (Illumina, followed by variant annotation and analysis using VarSeq (Golden Helix, Bozeman, MT, USA).
3. Case Report
3.1. Clinical Description
We investigated the genetic cause of cerebral malformations with unfavorable prognosis in a male fetus at 14 weeks of gestation.
Fetal ultrasound examination at the time of first-trimester scan revealed a severely disrupted cerebral and cerebellar development. The most striking anomaly was the presence of a large cranial encephalocele communicating with the right lateral ventricle (Figure 1A). The cerebral symmetry was disrupted, the left lateral ventricle was dilated, the thalamic area was hypoplastic, and the choroid plexus was small and shifted cranially and anteriorly (Figure 1B,C). The posterior fossa was missing the normal landmarks: the brain stem was hypoplastic, the fourth ventricle (intracranial translucency) could not be identified, the brain stem skull distance was severely enlarged, and the structures of the posterior fossa were replaced by a sonolucent area (Figure 1A,C). The maxillary cavities were prominent, unusual for this gestational age, and bilateral cleft palate could not be excluded (Figure 1D). No further anomalies of the fetus were identified.
The pregnancy was terminated in the 15th week of gestation due to the severity of the ultrasound findings.
3.2. DNA Analyses
SNP array analysis after chorionic villus sampling revealed a heterozygous de novo deletion in the chromosomal region 22q12.1 with a size of 58 kb (arr[GRCh38] 22q12.1(27710404x2, 27714323_27772264x1, 27772359x2)). The possibility of confined placental mosaicism was ruled out by also detecting the deletion in the DNA derived from fetal tissue after medically indicated termination of pregnancy. The deletion breakpoints were each located in intron 1 of the affected genes CPMER (lncRNA) and MN1 (protein-coding), indicating a possible gene fusion on the negative DNA strand (Figure 2A).
Additional trio-whole-exome sequencing and chromosome analysis did not reveal any evidence of another potentially causative variant in the fetus.
3.3. RNA/cDNA Analyses
To verify whether the MN1-CPMER fusion gene is actually transcribed into RNA, cDNA analyses were performed. Sanger sequencing of the cDNA revealed a direct link between exon 1 of MN1 and exon 2 of CPMER (Figure 2B). In addition, two further direct junctions between the first MN1 exon and CPMER were detected in the cDNA: one sequence contained a short segment of intron 4, which merged into exon 5, and one sequence showed a direct junction to exon 5 of CPMER. The results of these qualitative RNA analyses suggest the alternative splicing of the fusion pre-mRNA (Figure 2C). Each of the RNA transcripts could be detected under both conditions, the inhibition of nonsense-mediated mRNA decay (NMD) and without NMD inhibition. No fusion transcripts were detected in controls.
In silico protein sequence alignment demonstrated that the translation of the three identified fusion transcripts led to shortened MN1 proteins lacking all amino acids encoded by exon 2 (Figure 2D). Such C-terminally truncated MN1 proteins have been reported to be the cause of the extremely rare MCTT syndrome [5,6].
In addition, quantitative cDNA analyses were performed to quantify the expression level of the MN1-CPMER fusion gene. The amount of MN1 exon 1 transcribed was set as a reference value, since this exon was expressed in both alleles (wild-type and gene fusion). The expression level of MN1 exon 2 was about half the level of exon 1 expression under standard conditions (NMD not inhibited), indicating an almost equal expression of wild-type allele and gene fusion allele (Figure 2E). The analysis under NMD inhibition does not allow any conclusions to be drawn in this respect, due to the relatively high standard deviation for exon 2. In addition, the expression of one fusion transcript (MN1 E1-CPMER E2) was also determined quantitatively. An expression level of less than 50% was observed under both conditions, which is consistent with the previous detection of several fusion transcripts and possible alternative splicing (Figure 2E). The remaining two fusion transcripts could not be quantitatively investigated due to the limited amount of primary sample.
3.4. Database and Literature Review
A review of the international databases and the literature revealed no evidence for the presence of a comparable deletion with breakpoints in intron 1 of MN1 and CPMER. However, a similar deletion in 22q12.1, which also removed exon 2 of MN1 but did not result in the formation of a fusion gene, was identified in the germline of an apparently healthy individual from the 1000 Genomes Project (Figure 2F) [12].
4. Discussion
In the present study, a 58 kb heterozygous de novo deletion in 22q12.1 affecting the protein-coding gene MN1 and the lncRNA CPMER was detected in a male fetus as the cause of severe cerebral malformations. Our findings demonstrate that the germline deletion results in an MN1-CPMER gene fusion and the expression of stable mRNAs (Figure 2A–C). Furthermore, we were able to show that the fusion allele is expressed at a similar level as the wild-type allele (Figure 2E). The results also reveal alternative splicing and the expression of several fusion transcripts.
The translation of these mRNAs is expected to result in truncated MN1 proteins lacking the last 60 C-terminal amino acids encoded by exon 2 (Figure 2D). Mak et al. and Miyake et al. have shown in their studies that these C-terminally truncated proteins cause MCTT syndrome via a gain-of-function effect [5,6]. Besides intellectual and motor developmental abnormalities, craniofacial features and distinctive findings on brain imaging are key characteristics of MCTT syndrome [4]. The findings observed in the fetus are in line with the brain abnormalities described in MCTT syndrome, although due to the early stage of fetal development and the fact that the cerebellum is not yet developed, definite conclusions cannot be drawn.
The results of the literature and database review also point to the expression of the MN1-CPMER fusion gene as the cause of the fetal brain malformations. The deletion reported in the study of Levy-Sakin et al., in an apparently healthy person, without serious phenotypic abnormalities, also affects the second exon of MN1. In contrast to the fetal deletion, the deletion detected in this person does not involve the CPMER gene, and thus no fusion gene is formed (Figure 2F) [12]. Instead, the deletion is more likely to result in a loss of MN1 function due to nonsense-mediated mRNA decay. Heterozygous loss-of-function mutations in MN1 are not associated with facial and brain abnormalities [4]. This provides further evidence that the deletion detected in the fetus does not result in a loss of function but rather in a gain-of-function effect mediated by the expression of C-terminally truncated MN1 proteins.
In addition, it has not yet been reported that mutations in CPMER, which encodes the lncRNA Cytoplasmic Mesoderm Regulator, could be associated with brain abnormalities. In their recent study, Lyu et al. found that CPMER promotes cardiomyocyte differentiation in mice and humans [13].
In somatic gene fusions of PCGs and lncRNAs, Guo et al. found that the fusion transcripts are subject to regulation by the promoter of the upstream PCG [3]. Accordingly, the promoter of the MN1 gene is associated with the regulation of the studied MN1-CPMER gene fusion. MN1 is expressed in many different tissues, with high expression also in the brain, especially in the cerebral cortex and basal ganglia [14,15]. Unfortunately, we were unable to perform protein expression analysis as fetal tissue samples were not available.
In summary, all the obtained results indicate that the de novo deletion in 22q12.1 and the subsequent expression of the MN1-CPMER fusion transcript are causative for the fetal brain malformations. To the best of our knowledge, this is the first study reporting a germline gene fusion of a PCG and an lncRNA gene that is associated with a functional protein and related severe phenotypic abnormalities.
As Sánchez-Marín et al. recently stated in their review article, research on gene fusions involving lncRNAs “is still in its infancy”, with current focus solely on somatic cancer cells [2]. The results of our study not only expand the mutation spectrum at the MN1 locus by deletions resulting in the expression of C-terminally truncated proteins but also have locus-independent relevance. With this study, we want to raise awareness in the genetic community of the possibility that lncRNA fusions may result in protein expression with high clinical significance in the germline as well. Studying protein-forming gene fusions involving genes that are not originally coding genes unlocks novel opportunities in the search for disease-causing alterations.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Annala M.J. Parker B.C. Zhang W. Nykter M. Fusion genes and their discovery using high throughput sequencing Cancer Lett.201334019220010.1016/j.canlet.2013.01.01123376639 PMC 3675181 · doi ↗ · pubmed ↗
- 2Sánchez-Marín D. Silva-Cázares M.B. Porras-Reyes F.I. García-Román R. Campos-Parra A.D. Breaking paradigms: Long non-coding RN As forming gene fusions with potential implications in cancer Genes Dis.20241110113610.1016/j.gendis.2023.10113638292185 PMC 10825296 · doi ↗ · pubmed ↗
- 3Guo M. Xiao Z.D. Dai Z. Zhu L. Lei H. Diao L.T. Xiong Y. The landscape of long noncoding RNA-involved and tumor-specific fusions across various cancers Nucleic Acids Res.202048126181263110.1093/nar/gkaa 111933275145 PMC 7736799 · doi ↗ · pubmed ↗
- 4Mak C.C.Y. Fung J.L.F. Lee M. Lin A.E. Amiel J. Doherty D. Gordon C.T. Chung B.H.Y. MN 1 C-Terminal Truncation Syndrome Gene Reviews Adam M.P. Feldman J. Mirzaa G.M. Pagon R.A. Wallace S.E. Amemiya A. University of Washington Seattle, WA, USA 202032790267 · pubmed ↗
- 5Mak C.C.Y. Doherty D. Lin A.E. Vegas N. Cho M.T. Viot G. Dimartino C. Weisfeld-Adams J.D. Lessel D. Joss S. MN 1 C-terminal truncation syndrome is a novel neurodevelopmental and craniofacial disorder with partial rhombencephalosynapsis Brain 2020143556810.1093/brain/awz 37931834374 PMC 7962909 · doi ↗ · pubmed ↗
- 6Miyake N. Takahashi H. Nakamura K. Isidor B. Hiraki Y. Koshimizu E. Shiina M. Sasaki K. Suzuki H. Abe R. Gain-of-Function MN 1 Truncation Variants Cause a Recognizable Syndrome with Craniofacial and Brain Abnormalities Am. J. Hum. Genet.2020106132510.1016/j.ajhg.2019.11.01131839203 PMC 7042485 · doi ↗ · pubmed ↗
- 7Gene [Internet] Bethesda (MD): National Library of Medicine (US), National Center for Biotechnology Information 2004 Available online: https://www.ncbi.nlm.nih.gov/gene/4330(accessed on 22 February 2025)
- 8Kent W.J. Sugnet C.W. Furey T.S. Roskin K.M. Pringle T.H. Zahler A.M. Haussler D. The human genome browser at UCSC Genome Res.200212996100610.1101/gr.22910212045153 PMC 186604 · doi ↗ · pubmed ↗