Phenotypic Spectrum of KATNIP-Associated Joubert Syndrome: Possible Association with Esophageal Atresia and Review of the Literature
Maria Giovanna Tedesco, Ilaria Donati, Chiara Romeo, Sara Dal Bo, Chiara Nardini, Anna Maria Innoceta, Giulia Parmeggiani, Anna Patanè, Claudio Graziano

TL;DR
This paper reports a rare case of Joubert syndrome caused by a KATNIP gene variant and expands the known clinical features, including esophageal atresia.
Contribution
The study expands the phenotypic spectrum of KATNIP-related Joubert syndrome and suggests a possible link to esophageal atresia.
Findings
A novel KATNIP variant was identified in a child with Joubert syndrome and esophageal atresia.
KATNIP-related cases share JS features but often lack MTS and polydactyly.
Pituitary abnormalities are common in KATNIP-related Joubert syndrome.
Abstract
Background: Joubert syndrome (JS) is a multi-systemic ciliopathy, characterized by intellectual disability and congenital anomalies involving the brain, kidney, heart, and eye. Even if clinical presentation is variable, most authors consider a brain abnormality known as the molar tooth sign (MTS) as mandatory for diagnosis. About 40 genes were identified to be associated with JS, usually with an autosomal recessive pattern. KATNIP variants represent a rare cause of JS; only six families were previously reported. Methods: We performed exome sequencing in a child with a syndromic phenotype, described the clinical features and molecular findings, and performed a review of the literature to identify known individuals with pathogenic variants in KATNIP, highlighting clinical characteristics and gene-phenotype correlations. Results: Using exome sequencing, we identified a homozygous novel…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
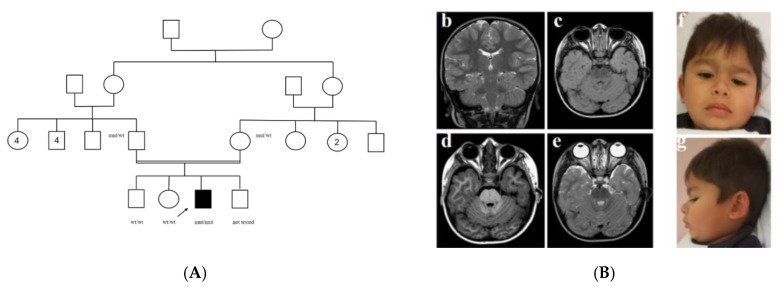 Figure 1
Figure 1Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsGenetic and Kidney Cyst Diseases · Hedgehog Signaling Pathway Studies · Genetic Syndromes and Imprinting
1. Introduction
Joubert syndrome (JS) is a multi-systemic ciliopathy that manifests early in life with hypotonia, developmental delay, abnormal eye movements (nystagmus, oculomotor apraxia), and respiratory pattern defects (apnea or tachypnea that may alternate). It is diagnosed by the presence of a peculiar cerebellar and brainstem malformation, known as the “molar tooth sign” (MTS) [1,2,3,4]. Intellectual disability is present in most, although a minority of individuals have normal cognition, and later involvement of other organs, such as the retina (chorioretinal coloboma, retinal dystrophy), kidney (cystic dysplasia evolving to nephronophthisis), liver (hepatic fibrosis), and skeleton (scoliosis, polydactyly), is frequent [5,6,7]. The molar tooth sign is usually considered pathognomonic and is related to the hypoplasia of cerebellar vermis and brain stem anomalies in axial plan on MRI [8,9,10]. More than 40 distinct genetic forms have been described, all following autosomal recessive inheritance (AHI1, CC2D2A, CEP290, CPLANE1, TMEM67 being the most frequently involved), except for X-linked OFD1 [2,11].
Autosomal recessive KATNIP-related JS was described for the first time in 2015 [12].
2. Materials and Methods
Here, we describe a male child with clinical features suggestive of a JS-related disorder, although he had esophageal atresia with tracheoesophageal fistula (EA/TEF) and a normal cerebral MRI without the molar tooth sign. Whole-exome sequencing (WES) identified a homozygous KATNIP pathogenic variant. WES was performed at R&I Genetics (Padova, Italy) on the NEXTSeq2000 platform (Illumina, San Diego, CA, USA) using the SureSelect All Exon V6 (Agilent, Santa Clara, CA, USA). Data were analyzed with BWA-GATK and proprietary pipelines, filtering for 1485 genes involved in neurodevelopmental disorders. Because of the result, the analysis was then extended to verify that there were no other significant variations.
The proband is the third child of consanguineous parents (Figure 1), who are first cousins of Roma ethnic background. Fetal ultrasounds were not performed regularly during pregnancy because of poor parental compliance. At birth, he showed type C esophageal atresia, which was surgically corrected on the second day of life but necessitated subsequent esophageal dilatation because of recurrence. Further congenital anomalies were bilateral post-axial polydactyly of the hands and an atrial septal defect, which resolved spontaneously. Rectal biopsies were performed for chronic constipation and possible Hirschsprung’s disease, but results were inconclusive. From the fifth month of life, he showed few episodes of flexion spasms and tonic-clonic seizures and was on anti-epileptic therapy for two years (no further episodes were reported). Cerebral MRI was performed, but no major parenchymal anomalies were detected except for slight diffuse enlargement of ventricular and periencephalic spaces. He had severe iron deficiency anemia, which required transfusion and was not responsive to martial supplementation.
He showed severe developmental delay, with absent language. At three years of age, he was 90 cm tall (3° centile), OFC was 47 cm (3° centile), weight 12.7 kg (3–10° centile), facial features were characterized by thick lips, cupped ears, anteverted nares, wide and sparse eyebrows (Figure 1). Ambulation was possible at four years, but only with support. At 5 years of age, a new clinical evaluation was performed: speech was absent, the same facial features were noted, he had a stature of 97 cm (<3° centile), weight was 15 kg (3° centile), and OFC was 48 cm (10–25° centile).
3. Results
Blood karyotype and Array-CGH were required as first tests, but no significant copy number variants were detected. Furthermore, IRIDA syndrome was suspected, based on unresponsive iron deficiency, but sequencing analysis of TMPRSS6 did not detect any pathogenic variants.
Afterwards, Whole Exome Sequencing was performed and filtered for genes associated with neurodevelopmental disorders. A c.808del (p.Ser270ValfsTer28) pathogenic variant in KIAA0556 (KATNIP, NM_015202.3) was identified in a homozygous state in the proband, whereas both parents were heterozygous. No other pathogenic/likely pathogenic variants were reported. Segregation analysis of the KATNIP variant was extended to the older healthy brother and sister, and it was absent in both (Figure 1). We suggested ENT examination with polysomnography and an endocrinology evaluation for the short stature, but the parents refused the evaluations. The MRI was reevaluated, but no signs suggestive of JS could be observed (Figure 1). Ophthalmological evaluation could not be completed due to a lack of compliance.
4. Discussion
KATNIP pathogenic variants represent a rare cause of JS (Joubert Syndrome 26 OMIM #616784) [12,13,14,15,16,17]. KATNIP protein binds directly to cytoplasmic microtubules and increases their stabilization; in C. elegans, disruption of the Kiaa0556 ortholog resulted in normal ciliary structure, function, and transport, although significant microtubule defects were detected by ultrastructural analysis [12]. To the best of our knowledge, 11 individuals (eight males, three females) from seven independent families are currently reported (Table 1).
Enrichment of consanguineous marriages is expected for ultrarare recessive disorders and parents were consanguineous in 5/7 KATNIP families. Data should be taken with caution from such a small cohort, but KATNIP–related JS seems neurologically milder than other forms of JS. Three of 11 individuals nevertheless had a severe developmental delay: in at least two of these families, the clinical phenotype is blended due to the presence of a 2nd variant [14,16] and expected to be more complex due to the additive effect of distinct variants. One additional variant is a homozygous loss-of-function variant in ADGRG1 [14], which is associated with complex brain malformations in humans [18]. In a second family [16], an additional variant of uncertain significance was reported in KIF7, which is a ciliopathy gene associated with variable phenotypes (including Joubert-12) [19] and is a strong candidate for disease modification. The third individual with a severe neurological phenotype is the proband of the current report, but we did not identify additional single-nucleotide or copy number variants involving genes that could explain a modification/worsening of the phenotype. KATNIP–related JS shows further peculiarities. Polydactyly, which is frequent in JS and a classical “ciliopathy” feature, was never reported before in individuals with KATNIP variants. Pituitary involvement is infrequent in other forms of JS but is clearly emerging as a specific feature of KATNIP-related JS: pituitary defects were described on brain MRI in at least 4/11, and endocrine abnormalities are common (three probands with proven growth hormone deficiency, one also had central hypothyroidism). Most individuals had short stature. Kidney abnormalities, which are frequent in JS, were never reported, but genital anomalies were common in males: 3/8 had cryptorchidism and/or micropenis, possibly due to pituitary hormone deficiency. On brain imaging, abnormalities of the corpus callosum and cerebellar hypoplasia were present (4/11 and 10/11, respectively), but four of 11 individuals did not show typical MTS. Furthermore, the proband of the current report showed severe unexplained anemia and EA/TEF. Iron deficiency anemia is often a multifactorial disorder, and the complex malformative/functional digestive issues of this child may be contributing factors; a hypothesis of IRIDA Syndrome was not confirmed by sequencing analysis of TMPRSS6. EA/TEF is a rare birth defect. It can be isolated, but it occurs within the context of additional anomalies in approximately 50% of individuals. It is a common feature of the VACTERL association spectrum, for which a specific molecular etiology is not known. Less than 10% of syndromic EA/TEF are caused by de novo chromosomal aberrations [20], and a few monogenic syndromes are known as well (e.g., Feingold syndrome caused by MYCN pathogenic variants) [21]. A possible involvement of ciliopathies in EA development is suggested by animal models, specifically, esophageal and tracheal anomalies were observed in a ciliopathy-associated mouse model with an Ift172 hypomorphic variant [22]. Esophageal atresia is not a specific feature of JS, but it is very interesting to note that KATNIP was already reported as a candidate gene for the etiopathogenesis of this defect [23], although one single individual was reported, with a de novo missense variant and a poor clinical description (esophageal atresia, heart defect, cleft lip). Pituitary anomalies are common and may lead to genital abnormalities in males and short stature. MTS can be absent, making the clinical diagnosis of JS challenging. The neurological phenotype is often mild but can be worsened by additional genetic variants in other genes that could affect clinical variability, as in the case of the patients described by Niceta and Cauley [14,16]. Thus, it is possible that further pathogenic variants, undetected due to technical limitations (e.g., small copy number variants or deep intronic variants), could modulate/influence the phenotype of this proband. Furthermore, the background genetic architecture could be studied through the implementation of polygenic risk scores and may act as a modifier of monogenic disorders [24]. Genome sequencing, which is now moving into clinical practice, would enable the analysis of intronic and regulatory regions and may soon help to determine the prognosis of rare disorders with better precision.
5. Conclusions
We describe the clinical features of a new proband with a homozygous loss-of-function KATNIP variant and compare these findings with the phenotype of a few previously reported individuals. Considering that our proband showed severe developmental delay and other rare features such as anemia unresponsive to iron supplementation and EA/TEF, it is intriguing to speculate about an undetected pathogenic variant (Deep intronic? Small copy number?) or an unfavorable genetic background. Nevertheless, we provide further evidence that ciliopathies contribute to esophageal atresia and identify KATNIP variants as an EA/TEF possible predisposing factor.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Alorainy I.A. Sabir S. Seidahmed M.Z. Farooqu H.A. Salih M.A. Brain stem and cerebellar findings in Joubert syndrome J. Comput. Assist. Tomogr.20063011612110.1097/01.rct.0000191681.05473.1316365585 · doi ↗ · pubmed ↗
- 2Gana S. Serpieri V. Valente E.M. Genotype-phenotype correlates in Joubert syndrome: A review Am. J. Med. Genet. C Semin. Med. Genet.2022190728810.1002/ajmg.c.3196335238134 PMC 9314610 · doi ↗ · pubmed ↗
- 3Sattar S. Gleeson J.G. The ciliopathies in neuronal development: A clinical approach to investigation of Joubert syndrome and Joubert syndrome-related disorders Dev. Med. Child. Neurol.20115379379810.1111/j.1469-8749.2011.04021.x 21679365 PMC 3984879 · doi ↗ · pubmed ↗
- 4Surisetti B.K. Holla V.V. Prasad S. Neeraja K. Kamble N. Yadav R. Pal P.K. Clinical and Imaging Profile of Patients with Joubert Syndrome J. Mov. Disord.20211423123510.14802/jmd.2106634592808 PMC 8490194 · doi ↗ · pubmed ↗
- 5Brancati F. Dallapiccola B. Valente E.M. Joubert Syndrome and related disorders Orphanet J. Rare Dis.201052010.1186/1750-1172-5-2020615230 PMC 2913941 · doi ↗ · pubmed ↗
- 6Maria B.L. Hoang K.B. Tusa R.J. Mancuso A.A. Hamed L.M. Quisling R.G. Hove M.T. Fennell E.B. Booth-Jones M. Ringdahl D.M. “Joubert syndrome” revisited: Key ocular motor signs with magnetic resonance imaging correlation J. Child. Neurol.19971242343010.1177/0883073897012007039373798 · doi ↗ · pubmed ↗
- 7Parisi M.A. The molecular genetics of Joubert syndrome and related ciliopathies: The challenges of genetic and phenotypic heterogeneity Transl. Sci. Rare Dis.20194254910.3233/TRD-19004131763177 PMC 6864416 · doi ↗ · pubmed ↗
- 8Maria B.L. Quisling R.G. Rosainz L.C. Yachnis A.T. Gitten J. Dede D. Fennell E. Molar tooth sign in Joubert syndrome: Clinical, radiologic, and pathologic significance J. Child. Neurol.19991436837610.1177/08830738990140060510385844 · doi ↗ · pubmed ↗