Chromosomal scale assembly and functional annotation of the apicomplexan parasite Eimeria acervulina
Subodh K. Srivastava, Carolyn C. Parker, Peter C. Thompson, Matthew S. Tucker, Benjamin M. Rosenthal, Asis Khan, Matthew J. Valente, Mark C. Jenkins

TL;DR
This paper presents a high-quality genome assembly and gene annotation for Eimeria acervulina, a parasite that causes coccidiosis in chickens, to aid in drug and vaccine development.
Contribution
The study provides a chromosomal-scale genome assembly and functional annotation for Eimeria acervulina using advanced sequencing and bioinformatics methods.
Findings
The genome assembly is 52 Mb with 15 chromosomes and 99% BUSCO completeness.
7,621 genes were predicted, with 60.97% having Pfam functions and 25.74% having Gene Ontology terms.
Stage-specific transcriptome analysis identified 9,761 transcripts.
Abstract
Apicomplexan parasites are single-celled obligate intracellular eukaryotic organisms that cause significant animal and human disease and pose a substantial health and socioeconomic burden worldwide. Eimeria acervulina is one such parasite of chickens, representative of several Eimeria species causing coccidiosis disease. A complete assembly of the E. acervulina genome may help identify markers of drug-resistance and design recombinant vaccines. We sequenced E. acervulina APU1 strain using Oxford Nanopore Sequencing and Illumina technology in combination with a Hi-C (Omni-C) proximity linkage library and achieved a chromosomal scale assembly using the MaSuRCA assembler. The final assembly was 52 Mb. with 15 chromosomes and 99% BUSCO completeness. A total of 7,621 genes were predicted using a pipeline of BRAKER3, GeneMark-ETP and AUGUSTUS, of which 4,647 (60.97%) have a predicted Pfam…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
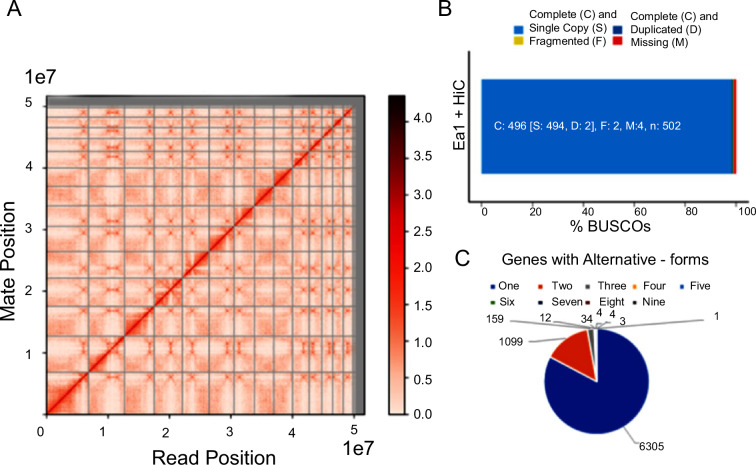 Figure 1
Figure 1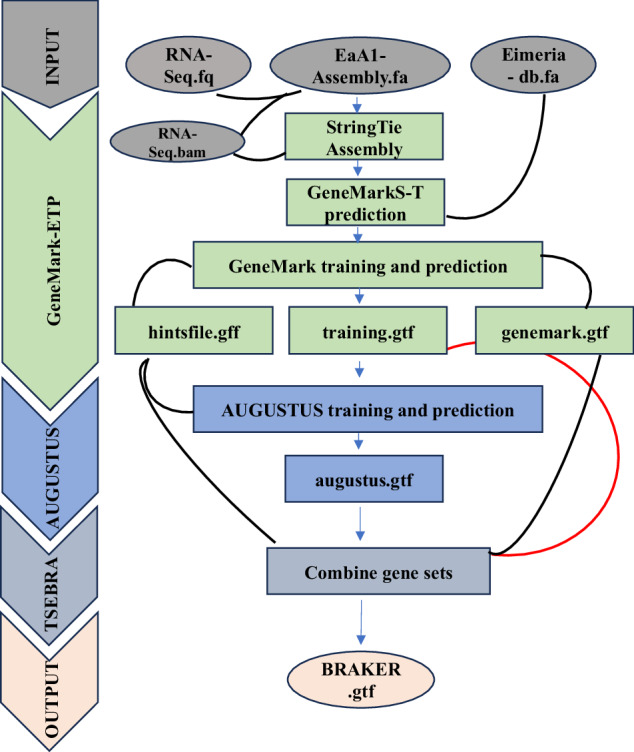 Figure 2
Figure 2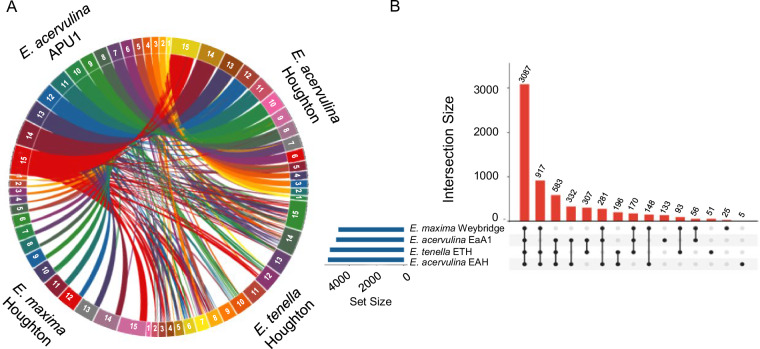 Figure 3
Figure 3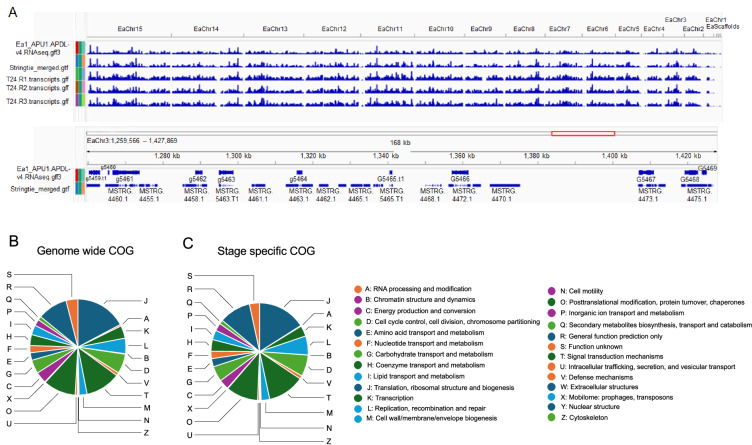 Figure 4
Figure 4Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsCoccidia and coccidiosis research · Parasitic Infections and Diagnostics · Helminth infection and control
Background & Summary
Apicomplexan parasites in the genus Eimeria cause enteric disease in livestock. Among livestock hosts, 12 Eimeria spp. have been described from cattle, 11 species from sheep, 9 from goats, 7 from turkeys and 7 from chickens^1,2^. Avian coccidiosis, widespread in poultry, causes more than $14 billion in economic damage each year to the poultry industry, worldwide^3,4^. This parasite invades epithelial tissues of the intestine, causing severe damage in birds and predisposes them to a potentially fatal infection called necrotic enteritis, leading to significant economic losses^5^.
Eimeria acervulina shares close similarity with other apicomplexan species that impair human and animal health^6^. In addition to its importance in chicken husbandry, it has utility as a surrogate organism for human parasites in the genus Cyclospora for basic and applied research applications^7–10^, addressing longstanding research gaps in less easily studied species^7^. Consequently, we sought a chromosomal scale assembly of Eimeria acervulina to establish its genetic architecture and provide insights relevant to parasite control.
To achieve a high-quality assembly, we used multiple sequencing technologies and a hybrid assembly approach. We used three different technologies to assemble this genome at chromosomal scale using Oxford Nanopore long reads (ONT using MinION™) (1,183,636 reads with a mean read length of 5k), Illumina NextSeq. 2000 short reads (78 million reads) and 18 million reads of Hi-C data (Omni-C) (Table 1). The hybrid mode MaSuRCA^11^ assembler produced contiguous sequences of 51.70 Mb containing 7,621 genes. The chromosomal scale assembly (Fig. 1A) was achieved by scaffolding with approximately 30X Hi-C sequence coverage produced on an Illumina sequencer and analysed with HiRise^12^.Table 1. Summary and type of obtained sequencing data generated and used in assembly and annotation of E. acervulina APU1.Sequencing librariesTotal dataMean Read Length (bp)Coverage (X)Nanopore (ONT)1,183,6365249120HiSeq-2000 PE78 M150250Hi-C (OminiC) PE17.8 M15030RNA-Seq50 M75—Fig. 1(A) Hi-C interaction matrix of de novo assembly contigs and Dovetail OmniC read pairs. Scaffolding was based on a likelihood model for genomic distance between read pairs which was used to identify and break putative misjoins, to score prospective joins, and make joins scaffold into chromosomes. (B) BUSCO analysis showed 99% completeness with 496 gene-sets out of 502 groups identified. Of these, 494 genes were complete and single copy, 2 complete and duplicated and, 2 fragmented and 4 missing genes. (C) Predicted genes with RNA-Seq shows 7,621 genes in E. acervulina APU1 with 1,316 genes showing alternative forms. (D) The Cluster of Orthologous Gene (COG) analysis found that 30.12% (1,899/6,305) single-copy genes have functional categories.
The genome completeness and quality were evaluated with Benchmarking Universal Single-Copy Orthologs (BUSCO), revealing 98.8% completeness (496) based on the 502 coccidia orthoDB v10 ortholog set. Of these, 494 genes were complete and single copy, 2 complete and duplicated, 2 fragmented, and 4 missing^13,14^ (Fig. 1B). Further analysis of gene prediction and annotation uncovered 7,621 genes with 6,305 primary genes and 1,316 designated as alternative forms^15^ (Fig. 1C). Annotation based on the Pfam v35.0 database with 19,633 entries and found 4,647 genes with Pfam functions and gene ontology (GO) terms with 1,962 classified into molecular, biological, and functional groups^16,17^. The best assemblies can include regions that have evolved through adaptations in paralogous sequences created by duplication. We compared our assembly to the previously published Eimeria acervulina Houghton assembly, which reported a 45.6 Mb genome comprising 3,415 scaffolds and 6,867 genes^18^. Our assembly increases the number of predicted genes by 11% and the total chromosomal assembly length by 12%, adding 6 Mb. The gene-to-gene comparison revealed 1,729 unique to the new APU1 assembly; 5,892 (77%) matched candidates in each isolate; their functional predictions are presented in Table S1.
This chromosomal scale assembly forms the basis for additional studies and facilitates an understanding of chromosomal rearrangement and genome structure. Furthermore, fine variations of gene and non-genic sequences may be examined for molecular, biological and functional differences that are involved in metabolism, growth and development of these organisms. Comprehending the genome biology of E. acervulina supports development of new drugs and vaccines to improve global food security^18^. We anticipate that this blueprint of chromosomal scale genome and functional analysis will hasten development of cost-effective vaccine candidates and facilitate biological research aimed at understanding parasite prevalence and preventing and controlling E. acervulina infections.
Methods
Sample collection and sequencing
E. acervulina oocysts were collected from litter on a commercial broiler farm in Maryland, USA and cloned by limiting dilution. These oocysts have since been propagated, as the APU1 strain, every 3-4 months in chickens at APDL, USDA, ARS, Beltsville for over 35 years. For DNA extraction, Oocysts are sporulated using standard procedures followed by storage in 2% K_2_CrO_4_ at 4°C. DNA extraction and preparation of sequencing libraries were carried out using methods developed in our laboratory^3^. Eimeria acervulina sporozoites were excysted from sporocysts that were released by grinding oocysts^19,20^. The sporozoites were centrifuged at 4000 rpm (2100 g) for 10 minutes, washed with saline A (0.14 M NaCl, 5 mM KCl, 4.2 mM NaHCO_3_, 0.1% glucose, pH 7.0), resuspended in 500 μl InhibitEX Buffer prior to DNA extraction using the QIAmp Fast DNA Stool Mini Kit according to manufacturer’s instructions (Qiagen, Germantown, MD). The integrity of DNA was analysed with Genomic DNA Screen Tape on a Tape Station instrument (Agilent Technologies, Santa Clara, CA) showing a DNA Integrity Number of 6.3 with peak size of 11.1 kb.
We first sequenced the genome of this isolate via long-read sequencing (Oxford Nanopore Technologies (ONT), Oxford UK) using 1 µg of genomic DNA and the ONT ligation sequencing kit SQK-LSK110. Approximately 200 ng of total library was sequenced for 48 h on a MinION flow cell (R9.4.1) per manufacturer recommendations^3,21^. To collect an adequate number of sequences, we repeated the sequencing twice and collected approximately 7 Gb of long read sequence data. We used Nanoplot and NanoStat software to evaluate the quality of sequencing results^22,23^. The sequence basecalling, translating raw electrical signal of Nanopore Sequencing to nucleotide sequence, influence the quality of sequences produced by Oxford Nanopore Technologies (ONT)^24^. We used the Guppy basecaller with the high accuracy model for basecalling purposes^24^. A complementary short-read dataset was produced on an Illumina NextSeq, employing an Illumina DNA Prep kit (Illumina, USA) with dual-indexed paired end Illumina indexes for sample identification^3^. Thirdly, contiguity was improved by interrogating chromatin interactions among physically proximate portions of the genome using Hi-C technology (Omni-C, CantataBio, Scotts Valley, CA)^3,25,26^.
To construct a library for proximity mapping using Omni-C, we excysted sporozoites in vitro and purified them using 5 µm PluriStrainer filters (Pluriselect, El Cajon, CA). The Hi-C library was prepared using the manufacturer’s instructions of their Dovetail Omni-C kit, version 2.0 (CantataBio, Scotts Valley, CA). Chromatin was fixed in place with formaldehyde in the nucleus and then extracted and digested with DNase I (at a further 1:10 dilution of the original Dovetail protocol for only 2 min instead of 30 min). Chromatin ends were repaired and ligated to a biotinylated bridge adapter followed by proximity ligation of adapter containing ends. After proximity ligation, crosslinks were reversed, and the DNA purified. The purified DNA was treated to remove biotin that was not internal to ligated fragments. Sequencing libraries were generated using NEB Next Ultra enzymes (New England Biolabs, Ipswich, MA) and Illumina-compatible adapters. Biotin-containing fragments were isolated using streptavidin beads before PCR enrichment of each library. The library was then sequenced on an Illumina HiSeqX platform to produce approximately 30X sequence coverage. Reads with mapping quality (MQ) greater than 50 were used for scaffolding (Fig. 1A).
Genome assembly and scaffolding with HiRise
We initially produced draft genome assemblies with the long reads derived from Oxford Nanopore Sequencing and the short reads derived from Illumina sequencing using the hybrid assembly capabilities of MaSuRCa. The draft assembly had 114 contigs with an N50 of 1,141,052 base pair (bp). Dovetail OmniC library sequences were aligned to the draft input assembly using bwa^27^. The separations of Dovetail OmniC with 13,518,859 read pairs mapped within draft scaffolds were analysed by HiRise. HiRise produced a likelihood model for genomic distance between read pairs, identified putative mis-joins, scored prospective joins, and ultimately made joins among contigs that scored above a threshold (weakspots -q 50 and export link -C16 resulted 50 scaffolds). The final chromosomal scale scaffolds were oriented using available chromosomal scale assembly of Eimeria tenella with reference-guided contig ordering and orienting in RaGOO and RagTag^3,28,29^.
Genome structure prediction and annotations
Gene prediction and annotation seeks to infer function from coding and non-coding regions of the genome. We used the BRAKER3 pipeline (Fig. 2) that uses GeneMark-ETP to integrate RNA-Seq and protein information to identify genes. The GeneMark predictions were fed into AUGUSTUS for gene model development and prediction; AUGUSTUS accuracy was improved with TSEBRA using RNA-Seq and orthologous genes^30^ as described in the workflow (Fig. 2). We analysed our chromosomal scale genome assembly with RNA-Seq data from E. acervulina^31^ and employing protein data from related species^5^. The software was trained to predict genes across the genome assembly with data from a combination of bulk transcriptomic libraries (approximately 50 million reads from E. acervulina APU1 sporulated oocysts at 24 hours) as described (Tables 1, 6)^31^ and Eimeria species that are known to infect chickens^30^.Fig. 2. Diagrammatic representation of E. acervulina APU1 genome prediction workflow using the BRAKER3 pipeline with RNA-Seq and annotated proteins from related Eimeria species. The BRAKER3 pipeline uses GeneMark-ETP to integrate RNA-Seq and protein information to identify genes. The Genemark results are then used by AUGUSTUS to generate gene models and predict genes ab initio; AUGUSTUS accuracy improves with TSEBRA using RNA-Seq data as described in workflow. The results of each step are combined to produce the final BRAKER gene annotations.
Data Records
The NCBI Bioproject for this chromosomal scale assembly can be found at PRJNA913161 at accession number JAVIVJ030000000^32^. Raw data for this genome assembly can be found in NCBI at SRA:SRP414521^33^ with SRR31596726, SRR31596727 and SRR31596728 as the individual run numbers. The transcriptome used in the study was from publicly available NCBI SRA accession number SRP324149^34^ with samples GSM5385954, GSM5385955 and GSM5385956.
Technical Validation
New sequencing technologies and chemistries enable more complete descriptions of complex genomes. Our E. acervulina genome assembly consists of 1.183 million high-quality long-reads (mean read length of 5,249 bases; mean quality score of 11.8) and 78 million high-quality NextSeq reads which were assembled by MaSuRCA^11^. This assembly produced an initial draft assembly of 51.70 Mb consisting of 114 contigs, N50 = 1,141,052 bases. We further constrained the assembly of these contigs employing Dovetail OmniC library reads of 17.5 Mb (~30 X coverage). Hi-C sequences were analysed by HiRise, to scaffold genome assemblies^12^ (Tables 1, 2). The improved Hi-C assembly provided 50 additional joins, and 2 breaks, to produced 50 scaffolds. These were re-scaffolded to fill gaps using evidence from ONT long reads, using recon and ntlink, to achieve chromosomal scale assembly. The longest scaffold thereby achieved was 6,840,217 bases, N = 50; 4,326,244 bases, L50 = 5. The shortest chromosomal scaffold was 845,591 bases. In total, 51,707,149 bases comprise our chromosomal scale genome assembly (Tables 2, 3)^35,36^. Assembly of mitochondrial and apicoplast organellar genomes was achieved from Nanopore and Illumina data employing the MitoHiFi^37^ and spades assemblers^38^, orientated with RaGOO and RagTag^28,29^. The assembled mitochondria and apicoplast genomes were 6,306 and 34,175 bp respectively (Table 3). These organelles were predicted and annotated to confirm the organelle specific genes.Table 2. Feature of Assembled E. acervulina APU1.AttributesAssembly featuresCountry of originUSASequencing TechnologyHybrid (ONT/NextSeq/Hi-C)Host /organismChicken/ParasitesGenome size (Mb)52Chromosomes15Longest chr sequence6,840,217Shortest chr sequence845,591N504,326,244L505Genome GC content (%)48.15Number of putative coding sequences7,621BUSCO Completeness; complete %99%CEG completeness: complete %80%Table 3. Statistics of E. acervulina APU1 genome length at chromosomal level.ChromosomeSizeChr156,840,217Chr145,836,873Chr134,856,252Chr124,586,734Chr114,326,244Chr104,059,355Chr93,320,098Chr83,179,687Chr73,001,917Chr62,688,218Chr52,090,460Chr41,751,870Chr31,705,884Chr21,535,592Chr1845,591Plastid; Apoplast34,175Mitochondria6,306Unplaced1,067,636Total51,733,109
We annotated the chromosomal scale genome assembly using multiple approaches, ultimately identifying 7,621 genes. Of these, 6,305 genes were represented exactly once in the genome; the remaining 1,316 appeared to have multiple forms showing close similarity with homologues from related species of Eimeria^18^ (Fig. 1C). The individual software combination of prediction was 4,365 by AUGUSTUS, 1,426 GMST and 514 GeneMark^39–41^ (Fig. 2). Pfam annotation was identified for 4,647 genes and the GO Gene Ontology database identified molecular, biological, and functional classes for 1,962. The Cluster of Orthologous Genes (COG) analysis revealed primary functional categories for 30.12% (1,899/6,305)^42^ (Fig. 1D).
Syntenic analysis and transcription factors
The syntenic relationship between E. acervulina APU1, E. acervulina Houghton, E. maxima Weybridge and E. tenella Houghton were analysed at the chromosomal level. We first constructed and orientated our chromosomal map to develop reference-guided contig ordering employing RaGOO^29^ and scaffolded these with RagTag^28^. We initially compared our assembly to those of the Houghton strain of E. tenella^26^, and with contig scale assemblies for the Houghton strains of E. maxima and E. acervulina^18^. We visualized contig synteny with Circa (http://omgenomics.com/circa) (Fig. 3A). As expected, the current assembly most closely matches that previously derived for the Houghton strain of E. acervulina^18^. We then examined evidence for collinearity among identified orthologous genes common to E. acervulina, E. maxima, and E. tenella^43^ (Fig. 3B). The comparative synteny between E. acervulina APU1 and E. acervulina Houghton was 44 Mb, whereas other Eimeria species were fragmented in small contig matches totalling 20 Mb with E. maxima and 32 Mb with E. tenella. We anticipate that fragmentation of syntenic regions could be driven by regions of high heterozygosity^3,18^.Fig. 3(A) Visualization of genome-wide synteny within (E. acervulina APU1 chromosomes vs E. acervulina Houghton) and between species (E. acervulina vs E. maxima vs. E. tenella) using Circa plot. E. acervulina APU1 and E. acervulina Houghton shared large genomic regions, whereas E. maxima and E. tenella were quite distant with only small, fragmented syntenic sections of their genomes. (B) UpSet analysis visualizes shared orthologs across groups of assemblies. Most orthologs were shared among all species examined. The E. acervulina assembly presented here had the highest number of unique orthologs of any assembly examined.
We identified transcription factors that may control growth and developmental pathways. Transcription factors (TF) are key regulators of gene expression that bridge cell signalling and gene regulation^44^. TF such as the MYB (essential for cellular growth) and AP2 (ApiAP2, which plays a crucial role in regulating various stages of parasite life cycle transitions by controlling gene expression at different developmental stages) are involved in the regulation of key processes during parasite development and stage transformation^45^. Transcription factors orchestrate many signalling pathways in eukaryotes, and aberrant transcription factors underly numerous human diseases^46^. Transcription factors also regulate parasite gene expression^47^. We analysed the E. acervulina chromosomes to identify TFs based on similarities to documented TFs in the Animal TFDB database^48^. The predicted proteins were analysed with BLASTp, searching the TF database which consists of 270k TFs from 182 animal genomes and are classified into 73 families. We found a total of 43 TF when querying the entire dataset (Table 4), and used those defined for Caenorhabditis elegans (C. elegans) to annotate E. acervulina TF candidates. Doing so identified 21 TF, which agrees with 590 members that list these candidates and their annotation using Pfam terms (Table 5).Table 4. The 43 Transcription Factor analysis using E. acervulina APU1 predicted proteins with database consists of large database of 270k TFs from 182 animal genome prediction and classified into 73 families.SL. No.EaA1 GenesFamilyfull Sequence e-valueBest domain e-valueDomain Number1g90.t1Germ_Cell_Nuclear_Factor-like0.0000710.00007162g206.t1SNF2-rel_dom1.00E-701.50E-7013g249.t1TRAM_LAG1_CLN84.30E-406.90E-4014g370.t1zf-C2H28.30E-300.0000395g398.t1BTB6.40E-163.90E-1526g432.t1SNF2-rel_dom7.10E-757.10E-7537g593.t1CSD1.80E-232.70E-2318g650.t1GCFC2.20E-322.20E-3239g655.t1MOZ_SAS7.00E-729.90E-72110g755.t1Myb_DNA-binding1.20E-112.30E-11111g1048.t1Tub5.90E-222.40E-15512g1136.t1Myb_DNA-binding0.00000370.0000077113g1384.t1NFYB4.80E-078.00E-07114g1418.t1Germ_Cell_Nuclear_Factor-like0.000020.000029115g1470.t1Germ_Cell_Nuclear_Factor-like0.0000470.006216g1474.t1SNF2-rel_dom1.20E-641.20E-64217g2069.t1zf-MIZ4.80E-174.80E-17218g2139.t1zf-LITAF-like8.10E-158.10E-15219g2361.t1HMG_box0.0000820.000082220g2464.t1HMG_box6.70E-080.00014221g2489.t1Myb_DNA-binding2.10E-171.20E-08222g2525.t1Myb_DNA-binding3.90E-111.00E-10123g2649.t1zf-C2H23.10E-080.0018224g2831.t1zf-C2H20.00000390.0000075125g3029.t1Myb_DNA-binding2.30E-186.80E-12226g3100.t1BTB7.70E-141.50E-13127g3181.t1HMG_box6.80E-221.00E-21128g3295.t1Myb_DNA-binding5.10E-101.10E-09129g3794.t1HMG_box5.60E-151.80E-14130g3800.t1SNF2-rel_dom4.00E-641.70E-63131g3941.t1HMG_box3.20E-081.00E-07132g3980.t1Germ_Cell_Nuclear_Factor-like0.0000990.00012133g4157.t1Myb_DNA-binding1.60E-080.0031334g4235.t1TRAM_LAG1_CLN84.60E-236.80E-23135g4348.t1SNF2-rel_dom4.60E-614.60E-61336g4499.t1MOZ_SAS1.20E-751.80E-75137g4832.t1Germ_Cell_Nuclear_Factor-like0.00000430.0000043238g4910.t1Myb_DNA-binding0.000170.00035139g5142.t1HMG_box1.10E-191.60E-19140g5175.t1BTB9.30E-070.0000025141g5200.t1SNF2-rel_dom1.40E-711.40E-71242g5211.t1BTB1.50E-143.80E-14143g5747.t1zf-MIZ2.60E-070.0000363Table 5E. acervulina APU1 Transcription factor annotations using Caenorhabditis elegans (C. elegans) genome (closest worm) available in the database to annotate E. acervulina TF candidates.SL. No.EaA1 genesC. elegans TF-genesTFPfam annotation1g15.t1ZK1067.2b.1ZK1067.2NA2g370.t1Y55F3AM.14.1zf-C2H2zf-C2H2 Zinc finger, C2H2 type3g593.t1F02E9.2a.1lin-28 CSDCSD ‘Cold-shock’ DNA-binding domain4g668.t1F40G9.17.1F40G9.17ACBP Acyl CoA binding protein5g755.t1Y113G7B.23 c.1swsn-1 MYBSWIRM SWIRM domain6g1152.t1F49E10.5a.1ctbp-1 THAP2-Hacid_dh_C D-isomer specific 2-hydroxyacid dehydrogenase, NAD binding domain7g1603.t1Y37E3.9.1phb-1 OthersBand_7 SPFH domain/Band 7 family8g2069.t1W10D5.3 c.1gei-17 zf-MIZzf-MIZ MIZ/SP-RING zinc finger9g2201.t1C32F10.5.1hmg-3 HMGRtt106 Histone chaperone Rttp106-like10g2226.t1F25H2.5.1ndk-1 OthersNDK Nucleoside diphosphate kinase11g2489.t1F38A5.13.1dnj-11 MYBDnaJ DnaJ domain12g3029.t1D1081.8.1cdc-5L MYBMyb_Cef pre-mRNA splicing factor component13g3181.t1Y48B6A.14.1hmg-1.1 HMGHMG_box HMG (high mobility group) box14g3794.t1F40E10.2.1sox-3 HMGHMG_box HMG (high mobility group) box15g3984.t1ZK783.4.1baz-2 MBDPHD PHD-finger16g4348.t1Y116A8C.13a.1SNF2SNF2-rel_dom SNF2-related domain17g4447.t1R05D3.11.1met-2 MBDSET SET domain18g4556.t1C16A3.4.1zf-C2H2zf-C2H2_2 C2H2 type zinc-finger (2 copies)19g4575.t1Y71G12A.3a.1tub-2 TubANAPC4_WD40 Anaphase-promoting complex subunit 4 WD40 domain20g5142.t1F47D12.4a.1HMGHMG_box HMG (high mobility group) box21g5927.t1F54F2.9.1MYBDnaJ DnaJ domain
Stage-specific novel gene prediction and annotation
Eimeria oocysts undergo sporulation (maturation to the infectious stage by developing sporozoites in structures called sporocysts) under the influence of temperature, oxygen, and moisture^6,49^. The stage-specific transcriptomic profile shed light on the drivers and consequences of developmental change^50,51^. Chromosomal scale assembly supported by transcriptome profiles aid in genetic mapping and identifying transcriptional and post-transcriptional variations and Quantitative Traits Loci (QTL)^52^. We therefore supported gene annotation using transcriptome libraries produced from three biological replicates of oocysts of our sequenced strain of E. acervulina (APU1) as they underwent maturation for 24 hours^31^. Each biological replicate had over 50 million paired end reads (Table 6).Table 6E. acervulina APU1 sporulation, stage specific (T24) novel gene prediction and annotation data (RNA-Seq) biological replicates statistics used in the analysis.RNA-SeqLeft readsRight readsOverall read mapping rateT24_rep157,344,16257,344,16294.30%T24_rep234,319,24434,319,24494.50%T24_rep354,059,75554,059,75593.20%Avg.48,57438748,574,38794.00%
The total transcriptomes revealed 47,556 exons in 9,761 transcripts. There were multiple transcripts for 3,601 of these loci which averaged 2.9 transcripts per locus. We compared the BRAKER3 predicted 7,621 genes from the assembled genome to the stage specific transcriptome assembly from the Cufflinks pipeline^53^. 44% (4,293) of Cufflinks and merged with transcripts with Stringtie^54^ compared common with BRAKER3 predictions. The 56% (5,468) of transcripts without BRAKER3 pipeline support due to reference-guided prediction (Table S2). The additional transcripts predicted from read-support (ORFs in RNA-Seq assemblies) were called cufflinks predicted novel transcripts^31,55^. These assembly transcripts were analysed for ORFs to find coding regions within transcripts (TransDecoder) with the minimum length criteria of 50 amino acids^56^. We retrieved 9,325 transcripts followed by Pfam and COG analysis (Table 5). Out of these 7,054 genes, Pfam identified with 4,054 having BRAKER3 annotation-supported genes and an additional 3,000 transcripts derived from de novo assembly of the sporozoite transcriptome (Fig. 4A and Table S2). The COG analyses classified 31.64% (2,951/9,325) sequences into COG functional categories (Fig. 4B,C and Table 7). These transcriptomes consider stage-specific transcripts (novel) associated with sporulation at 24 hours and analysed gene annotation and functional categories. The genome-wide transcriptome profile is depicted in the top view (Fig. 4A). The bottom view identifies stage-specific (novel) transcripts not found in the gene prediction and chromosomal scale genome, as judged by a multi-criterion approach for gene prediction and annotation; it uncovered 7,621 genes (Fig. 4B). Cluster of Orthologs (COG) analysis reveals that 30.12% (1,899/6,305) primary genes have functional categories. The stage-specific transcriptome assembly (T24) retrieved 9,325 genic-level transcripts out of 31.64% (2,951/9,325) sequences classified into COG functional category (Fig. 4C). These novel transcripts were annotated to understand their function, enabling their use to characterize for protein expression and immune responses^57^. Further in-depth analysis of parasite transcription factors, especially those that regulate virulence, may identify novel drug targets. Manipulation of these TFs may elucidate gene expression at various stages and improve gene annotation. We mapped all predicted and sporulation assembled transcripts to the chromosomal coordinates to establish their locations (Table S2).Fig. 4(A) Comparison of gene density across the genome for BRAKER3 gene predictions in this assembly and transcripts from RNAseq assembly replicates of E. acervulina sporulation at 24 hours.(Top view). The bottom view highlights differences between sporulation stage-specific (novel) transcripts and gene prediction for the chromosomal genome assembly. (B) Chromosomal scale gene predictions using the BRAKER3 pipeline uncovered 7,621 genes. Cluster of Orthologous Gene (COG) analysis revealed that 30.12% (1,899/6,305) single-copy genes have functional categories. (C) Stage-specific transcriptome assembly (T24) produced 9,325 genic-level transcripts; 31.64% (2,951 /9,325) of sporulation-specific transcript sequences were classified into COG functional categories.Table 7E. acervulina APU1, E. acervulina Houghton and E. acervulina sporulated (T24) Transcriptomes assembly COG annotation variation.LETTEREaA1EAHSporulated RNA (T24)DESCRIPTIONA1096RNA processing and modificationB331Chromatin structure and dynamicsC736698Energy production and conversionD126144211Cell cycle control, cell division, chromosome partitioningE486080Amino acid transport and metabolismF463872Nucleotide transport and metabolismG88100152Carbohydrate transport and metabolismH7086114Coenzyme transport and metabolismI626394Lipid transport and metabolismJ327322473Translation, ribosomal structure and biogenesisK8187105TranscriptionL99142186Replication, recombination and repairM515384Cell wall/membrane/envelope biogenesisN647Cell motilityO208208315Posttranslational modification, protein turnover, chaperonesP415076Inorganic ion transport and metabolismQ243539Secondary metabolites biosynthesis, transport and catabolismR192199311General function prediction onlyS7671100Function unknownT225238362Signal transduction mechanismsU141217Intracellular trafficking, secretion, and vesicular transportV202733Defense mechanismsW000Extracellular structuresX425Mobilome: prophages, transposonsY000Nuclear structureZ5410Cytoskeleton
In summary, this high-quality chromosomal scale genome assembly, combined with transcriptome annotation, provide insights into the genome evolution of E. acervulina. Accomplishing this chromosome-level genome will enable future studies of chromosomal rearrangement, investigations of pervasive gain or loss of genomic content, hasten identification of Quantitative Trait Loci (QTLs), and facilitate functional and evolutionary analyses of genes. Chromosomal orientation will hasten fine mapping of genes and examination of variation among closely related species. These studies may contribute to drug discovery and knowledge of host-parasite interactions that can impact control of parasitic disease.
Supplementary information
Table S1. Table S2.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Marchiondo, A. A., Cruthers, L. R. & Fourie, J. J. Parasiticide screening: in vitro and in vivo tests with relevant parasite rearing and host infection/infestation methods. (Elsevier/Academic Press, 2019).
- 2Tucker, M. S., Khan, A., Jenkins, M. C., Dubey, J. P. & Rosenthal, B. M. Hastening Progress in Cyclospora Requires Studying Eimeria Surrogates. Microorganisms 10, 10.3390/microorganisms 10101977 (2022).10.3390/microorganisms 10101977 PMC 960877836296256 · doi ↗ · pubmed ↗
- 3Taha, S. et al. Interplay between and during In Vitro Infection of a Chicken Macrophage Cell Line (HD 11). Life-Basel 13, (2023).10.3390/life 13061267 PMC 1030147637374050 · doi ↗ · pubmed ↗
- 4De Coster, W. & Rademakers, R. Nano Pack 2: population-scale evaluation of long-read sequencing data. Bioinformatics 39, 10.1093/bioinformatics/btad 311 (2023).10.1093/bioinformatics/btad 311PMC 1019666437171891 · doi ↗ · pubmed ↗
- 5Gabriel, L. et al. BRAKER 3: Fully automated genome annotation using RNA-seq and protein evidence with Gene Mark-ETP, AUGUSTUS and TSEBRA. bio Rxiv, 10.1101/2023.06.10.544449 (2024).10.1101/gr.278090.123PMC 1121630838866550 · doi ↗ · pubmed ↗
- 6Bruna, T., Lomsadze, A. & Borodovsky, M. A new gene finding tool Gene Mark-ETP significantly improves the accuracy of automatic annotation of large eukaryotic genomes. bio Rxiv, 10.1101/2023.01.13.524024 (2024).10.1101/gr.278373.123PMC 1121631338866548 · doi ↗ · pubmed ↗
- 7Weidemuller, P., Kholmatov, M., Petsalaki, E. & Zaugg, J. B. Transcription factors: Bridge between cell signaling and gene regulation. Proteomics 21, (2021).10.1002/pmic.20200003434314098 · doi ↗ · pubmed ↗
- 8Wang, J. L. et al. The transcription factor AP 2XI-2 is a key negative regulator of merogony. Nature Communications 15, (2024).10.1038/s 41467-024-44967-z PMC 1081796638278808 · doi ↗ · pubmed ↗