Bone Marrow Infection by Pneumocystis jirovecii in a Patient with AIDS: A Case Report and Literature Review
Diego Alejandro Cubides-Diaz, Valentina Negrette-Lazaro, Viviana Poveda-Hurtado, Juan Pablo López-Salazar, Carlos Mauricio Calderón-Vargas, Carlos Arturo Álvarez-Moreno

TL;DR
A patient with AIDS developed a rare bone marrow infection caused by Pneumocystis jirovecii, highlighting the need for early suspicion and proper diagnosis in immunosuppressed individuals.
Contribution
This case report adds to the limited literature on extrapulmonary Pneumocystis jirovecii infection involving the bone marrow in advanced HIV/AIDS patients.
Findings
Bone marrow involvement by Pneumocystis jirovecii was confirmed via biopsy in a patient with AIDS.
The patient also had Kaposi sarcoma and died despite antimicrobial therapy.
Persistent cytopenias and systemic symptoms should raise suspicion for disseminated Pneumocystis infection.
Abstract
Background: Pneumocystis jirovecii primarily causes pneumonia in immunosuppressed individuals, particularly those living with advanced HIV/AIDS. Extrapulmonary dissemination is uncommon, with bone marrow involvement described in only a handful of cases globally. Bone marrow infection occurs in the setting of severe immunosuppression, poses diagnostic challenges, and carries a high mortality rate. Methods: We describe the case of a 34-year-old man newly diagnosed with HIV/AIDS, presenting with severe immunosuppression and Pneumocystis jirovecii pneumonia. The patient initially improved with cotrimoxazole and corticosteroids, but was readmitted shortly after discharge with abdominal pain, diarrhea, and worsening pancytopenia. A bone marrow biopsy revealed Pneumocystis jirovecii cysts, confirming disseminated infection. Concomitant Kaposi sarcoma involving the skin and gastrointestinal…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
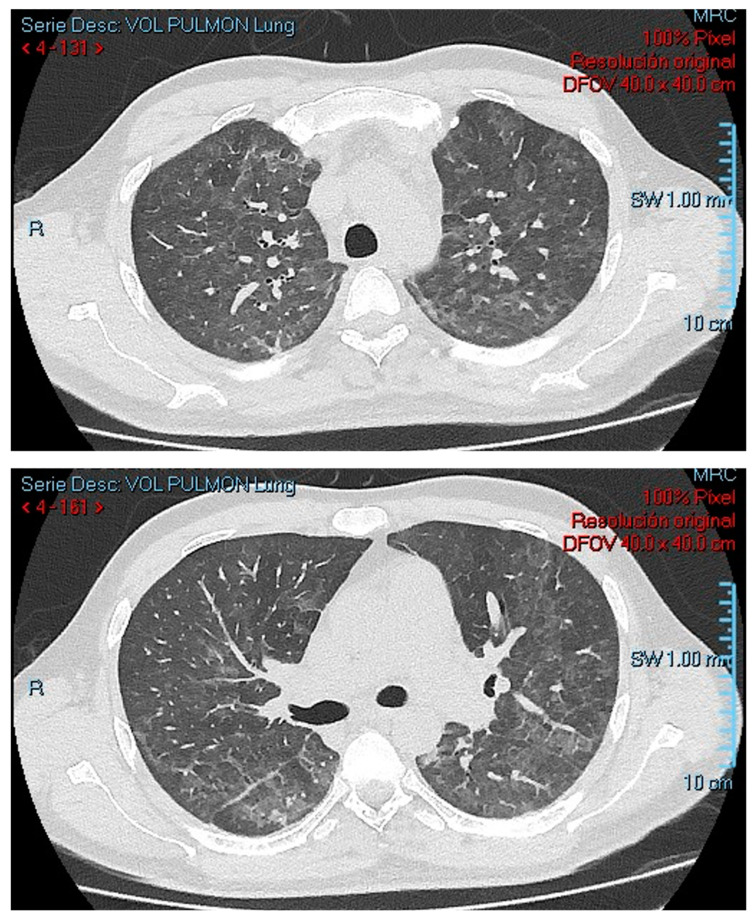 Figure 1
Figure 1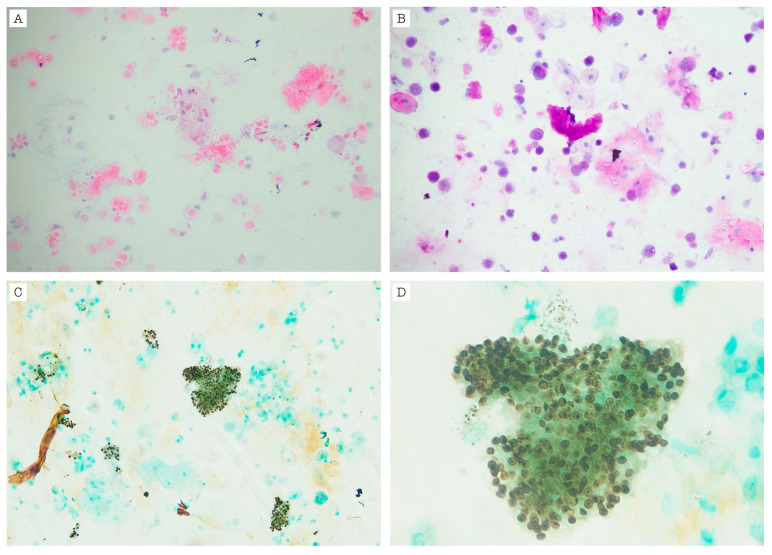 Figure 2
Figure 2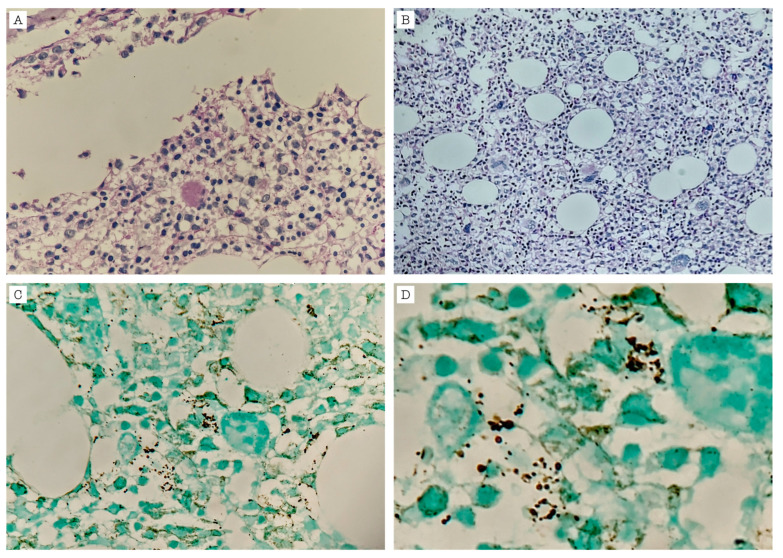 Figure 3
Figure 3Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsPneumocystis jirovecii pneumonia detection and treatment · HIV/AIDS drug development and treatment · Pneumonia and Respiratory Infections
1. Introduction
Pneumocystis jirovecii (P. jirovecii) is an obligate fungal pathogen with parasitic behavior that colonizes the human respiratory tract and is primarily transmitted through airborne human-to-human contact. P. jirovecii can cause life-threatening pneumonia in individuals with impaired immunity such as those with advanced HIV/AIDS (particularly with CD4 counts <200 cells/μL), hematologic malignancies, solid organ transplants, or those receiving immunosuppressive therapies [1]. P. jirovecii pneumonia (PCP)—still commonly referred to by the acronym PCP to maintain consistency across the literature—remains a major opportunistic infection among people living with HIV, but its impact is increasingly recognized in non-HIV immunocompromised populations [1,2]. Genotyping studies have demonstrated that more than 90% of PCP cases involve genetically distinct mixtures of P. jirovecii strains, supporting the hypothesis that ongoing exposure and repeated inhalation from diverse sources—rather than the reactivation of latent organisms—is the primary mechanism driving PCP pathogenesis [1].
In recent years, PCP has emerged as a critical concern among patients with hematologic malignancies, solid tumors, organ transplants, and those receiving immunosuppressive therapies [2]. Unlike the subacute clinical course typically observed in HIV-positive individuals, PCP in non-HIV patients presents with a rapid onset and progression to respiratory failure, often within days. These patients frequently experience more severe hypoxemia, and a higher mortality rate compared to their HIV-infected counterparts. Diagnostic challenges are more pronounced in the non-HIV population due to a lower fungal burden. Radiologic findings also differ, ranging from classical diffuse ground-glass opacities to more sharply demarcated panlobular patterns [2]. Therapeutically, cotrimoxazole remains the first-line treatment; however, the optimal dosing and duration in non-HIV patients are not fully standardized. The benefit of adjunctive corticosteroids in this population also remains controversial [2]. Extrapulmonary infection by P. jirovecii is rare, with the lymph nodes and spleen being the most frequently affected sites [3]. Bone marrow involvement has been described only in a limited number of cases worldwide [3,4,5,6,7,8,9,10,11,12,13]. The primary risk factor associated with bone marrow infection is HIV/AIDS, particularly in patients who have not received adequate pharmacologic prophylaxis for P. jirovecii, such as systemic cotrimoxazole or dapsone. Inhaled pentamidine, although once widely used, has been associated with an increased risk of extrapulmonary disease due to insufficient systemic levels [3,4]. This stage of infection is believed to represent hematogenous or lymphatic dissemination from a primary pulmonary focus and carries a poor prognosis, with mortality rates reported to exceed 90% in several series [3,4,5,6,7,8,9,10,11,12]. In the comprehensive review by Ng et al., most cases of disseminated P. jirovecii with bone marrow involvement occurred in patients with primary pulmonary disease and were diagnosed postmortem [13]. Diagnosis typically relies on histopathologic examination, and in many instances, the condition is identified incidentally during the evaluation of tissue samples [3,4,5,6,7,8,9,10,11,12,13].
On the other hand, Kaposi sarcoma (KS) is a vascular tumor associated with human herpesvirus 8 (HHV-8) in patients living with HIV. It typically affects the skin, mucosa, and lymph nodes, but can also present with visceral dissemination, particularly involving the lungs and gastrointestinal tract, which worsens the prognosis [14]. KS may also be associated with hyperinflammatory syndromes such as immune reconstitution inflammatory syndrome (KS-IRIS) and inflammatory cytokine syndrome (KICS). KS-IRIS can present either by exacerbating pre-existing lesions or unmasking subclinical disease, often leading to rapid clinical deterioration. Unlike IRIS caused by other opportunistic infections, corticosteroids should be avoided in KS-IRIS due to the risk of accelerating viral replication and tumor progression [14]. KICS, a more recently recognized condition, is characterized by systemic inflammatory response syndrome (SIRS) with high levels of HHV-8 viremia and elevated interleukins (IL-6 and IL-10). Clinically, KICS often overlaps with multicentric Castleman disease (MCD), although without histological confirmation of MCD. KICS carries a high mortality rate and may occur concurrently with KS-IRIS, as both conditions are driven by immune dysregulation in the setting of viral replication. Prompt recognition and differentiation between these KS-driven complications are essential to guide appropriate management [14].
We present the case of an adult living with HIV who developed PCP, which subsequently progressed to disseminated disease with bone marrow involvement. During hospitalization, a concurrent diagnosis of KS affecting the skin and gastrointestinal tract was established, further complicating the clinical course. A summary of the patient’s clinical presentation and laboratory findings is provided below.
2. Case Report
A 34-year-old man with a history of hypertension and unprotected sexual activity presented with a three-month history of fatigue, night sweats, an 11-pound involuntary weight loss, dry cough, intermittent non-dysenteric diarrhea, and progressive dyspnea. On examination, he was dehydrated, had an oxygen saturation of 78%, and exhibited oral lesions suggestive of candidiasis. Initial laboratories revealed mild thrombocytopenia, a positive four generation HIV test, reactive serologic tests for Treponema pallidum, and a blood arterial gas test with a moderate oxygenation disorder and a high alveolar–arterial gradient (Table 1). A chest X-ray showed left basal interstitial opacities, later characterized on chest tomography, revealing extensive ground-glass opacities randomly distributed in both lung fields (Figure 1).
The patient was considered to have a diagnosis of HIV infection, latent syphilis, oral candidiasis, and multilobar pneumonia of an undetermined etiology. The CD4 count was 33 cells/mm^3^, and the HIV viral load was 2,095,776 copies/mL. Due to the immunosuppressive condition and the tomographic pattern, a PCP was suspected. Antimicrobial therapy was started with fluconazole, benzathine penicillin, and cotrimoxazole with corticosteroids. The fibrobronchoscopy and bronchoalveolar lavage revealed structures suggestive of P. jirovecii (Figure 2), confirming the diagnosis of severe PCP and ruling out other infectious conditions. Therapy with cotrimoxazole was continued at a dose of 20 mg/kg/day, based on the trimethoprim component, as well as prednisolone on a tapering regimen. After 12 days of hospitalization, the patient was discharged on oral cotrimoxazole and prednisolone for 21 days, with plans to initiate antiretroviral therapy as an outpatient. The platelet count continued to be low, and other opportunistic infections, such as tuberculosis and histoplasmosis, were ruled out (Table 2).
Five days after discharge, the patient was readmitted for persistent abdominal pain, diarrhea, emesis, and fever. On readmission, laboratory tests showed persistent thrombocytopenia, leukocytosis, neutrophilia, elevated C-reactive protein, and hyponatremia (Table 1). Stool samples were negative for parasitic, bacterial, or fungal infection, and abdominal computed tomography revealed splenomegaly and an inflammatory thickening of the distal ileum.
During hospitalization, the patient developed pancytopenia, prompting a bone marrow biopsy, which revealed foamy, amorphous eosinophilic material and small, non-budding round structures suggestive of P. jirovecii yeasts (Figure 3), confirming disseminated infection with bone marrow involvement. A diagnosis of KS was confirmed by a histopathologic analysis of both a skin biopsy and an inguinal lymph node biopsy. Additionally, video capsule endoscopy revealed lesions consistent with KS involving the esophagus, duodenum, jejunum, and ileum. By that time, therapy with cotrimoxazole had been completed for a total of 21 days, with a progressive resolution of leukopenia and thrombocytopenia, and he was referred to an oncology institution to complete oncologic and antiretroviral treatment. Two months later, the patient was hospitalized again due to clinical deterioration, ultimately succumbing to multisystem organ failure.
3. Discussion
P. jirovecii infection presents in patients with immunosuppressive conditions such as AIDS, hematologic or solid organ malignancies, and iatrogenic immunosuppression [1,2]. This infection primarily affects the lungs, as the fungus adheres to and erodes type 1 pneumocytes, leading to diffuse alveolar damage [15]. In immunocompetent individuals, P. jirovecii colonization is typically asymptomatic and self-limited. This effective control is mediated through the coordinated activation of antigen-presenting cells, CD4+ T cells, and B cells within bronchus-associated lymphoid tissue. These structures facilitate communication between CD4+ T cells—particularly follicular helper T cells—and B cells, leading to M2 macrophage polarization and fungal clearance with minimal inflammation [16]. In contrast, immunocompromised hosts—especially those with CD4+ T cell depletion—fail to mount this organized response. The fungus proliferates in the alveoli, triggering a poorly regulated, pro-inflammatory reaction. In this context, CD8+ cytotoxic T cells, NK cells, and pro-inflammatory Th1 responses predominate, promoting M1 macrophage activation through IFN-γ and TNF-α. While these pathways enhance fungal clearance, they also contribute significantly to pulmonary inflammation and tissue damage. Furthermore, P. jirovecii surface antigens such as major surface glycoprotein (Msg) and β-D-glucan stimulate Toll-like and C-type lectin receptors, enhancing local inflammation and impairing surfactant production, thus compromising alveolar integrity and gas exchange [16,17].
The most common symptoms are dry cough, progressive dyspnea, fever, and low exercise tolerance [15,18]. The incubation period from infection to the onset of clinical disease is approximately 5 to 6 days but sometimes can be extended up to 28 days [15]. Upon consultation, most patients present with hypoxemia, a high alveolar–arterial gradient, and in some cases, respiratory failure [15,18].
Diagnosing PCP requires a susceptible immunosuppressed host with a compatible clinical syndrome, and suggestive laboratory and imaging findings [19]. Chest computed tomography is the imaging modality of choice and usually reveals diffuse ground glass lung opacities, sometimes accompanied by lobar consolidations and subpleural compromise. In contrast, other less frequent tomographic findings, such as cysts or pneumothorax, can also be found [19,20]. Laboratory diagnosis usually relies on indirect methods such as a measurement of serum lactate dehydrogenase [LDH] which is elevated in most cases of PCP in people living with HIV, due to its release from damaged lung tissue [19,21]. A sensitivity and specificity of 86% and 45.3% have been described, with a cut-off point of 268 UI/L [19,21]. Because P. jirovecii cannot be cultured ex-vivo, diagnosis depends on a direct visualization of the fungus or molecular detection of its DNA [19,21]. Currently, the gold standard method is the analysis of bronchoalveolar lavage, with a reported sensitivity of 95–100% [15,19,21]. A diagnosis can be made with the observation of the trophic or cystic forms with special stains such as Giemsa, Wright, Diff-Quik, Cresyl violet or Grocott–Gömöri’s methenamine silver; however, a diagnostic can also be made by the presence of eosinophilic amorphous exudates in the alveolar spaces, which can be observed with routine staining [15,19,21]. Specimens obtained through other techniques such as expectorated sputum, or induced sputum have a lower and variable performance [19,21]. The molecular diagnosis of PCP is based on detecting several gene loci with polymerase chain reaction [PCR] techniques, and performance improves by using multicopy targets such as Msg, mtLSU, HSP70, and cdc2 [22]. The sensitivity of these techniques can be improved by using nested PCR (nPCR). Since the fungus can colonize the airways without establishing infection, the specificity can be also improved with the use of quantitative PCR (qPCR), which can be performed on invasive and non-invasive specimens [22]. In people living with HIV, the detection of HSP70 by qPCR on bronchoalveolar lavage reported a sensitivity and specificity of 98% and 96%, respectively [23], and the detection of Msg by qPCR on oral wash reported a sensitivity and specificity of 88% and 85%, respectively [24]. A negative PCR will likely exclude PCP, especially in deep respiratory samples [25], but a positive PCR requires clinical interpretation to distinguish between colonization and active infection, and there is still a need for more research to validate and establish specific cut-off points for these molecular techniques [19,22,25].
Extrapulmonary infection by P. jirovecii is rare and the manifestations depend on the compromised organ [3]. The most commonly affected sites are the lymph nodes, liver, spleen, gastrointestinal tract, and bone marrow; with 64% of these cases reporting multiple organ involvement [3]. Excluding the bone marrow, tomographic studies often reveal hypodense lesions with a tendency to calcify on the affected organs, which progress to rimmed or complete calcifications after successful treatment [3]. The primary risk factor for developing an extrapulmonary infection is the AIDS stage of HIV infection, especially those patients who do not receive adequate prophylaxis with cotrimoxazole or receive other pharmacological regimens such as inhaled pentamidine [3,4,9,10,12]. Other risk factors include hypogammaglobulinemia and hematologic malignancies [3,23].
The mechanism by which P. jirovecii reaches the bone marrow remains unclear. Almost all patients with bone marrow compromise had a pulmonary infection first, which strongly suggests that the fungus reaches the bloodstream from the lungs [3,5,6,7,8,10,11,12]. It is believed that in severely immunosuppressed hosts, the microorganism reaches the pulmonary lymph nodes, escapes phagocytosis, and disseminates into the venous bloodstream through the efferent lymphatic drainage [5]. Alternatively, some histologic evidence suggests a possible mechanism through alveolar septal destruction and direct capillary wall invasion [5]. Upon reaching the bloodstream, the bone marrow compromise may represent just a stage of a disseminated infection, like what happens in miliary tuberculosis [5,26].
To our knowledge, bone marrow infection by P. jirovecii has been reported in only a small number of cases worldwide (Table 3) [3,4,5,6,7,8,9,10,11,12]. In our review, 63.6% of the affected patients were living with AIDS [3,5,6,8,9,10,11,12], while the remainder had various forms of lymphoma [4,7]. The mean age was 42 years, and 81.8% of the cases occurred in male patients. [3,4,5,6,7,8,9,10,11,12]. The most frequent symptoms were dyspnea (45.4%) and fever (36.3%). Blood cytopenias such as anemia, leukopenia, and thrombocytopenia were described in almost all of these cases [3,4,5,7,8,10,12], as well as primary pulmonary infection, which supports the hypothesis that P. jirovecii reaches the bloodstream from the lungs through the efferent lymphatic drainage after escaping phagocytosis, or by a direct invasion of capillaries [3,5,6,7,8,10,11,12]. All these cases were diagnosed with a histologic examination of the bone marrow, and the most frequent findings were the presence of amorphous eosinophilic exudates or P. jirovecii cysts [3,4,5,6,7,8,10,11,12]. Treatment regimens were variable, and a high mortality rate greater than 90% was described despite treatment [3,4,5,6,7,8,9,10,11,12].
In this case, while the diagnosis of disseminated P. jirovecii infection was confirmed histologically through bone marrow biopsy, the subsequent clinical deterioration and death raise the possibility of additional or alternative diagnoses. One potential contributor is KS-IRIS, particularly if antiretroviral therapy was initiated after discharge. KS-IRIS is known to present with a rapid progression of KS lesions and systemic inflammatory features, particularly in the presence of pulmonary involvement, which carries a poor prognosis. Another possible diagnosis is KICS, a hyperinflammatory state driven by HHV-8 replication that often overlaps clinically with multicentric Castleman disease [14]. KICS is characterized by features such as pancytopenia, SIRS, and hyponatremia, all of which were present in this case. Additionally, hemophagocytic lymphohistiocytosis (HLH) must be considered. HLH is a hyperinflammatory syndrome that can occur in the setting of HIV, opportunistic infections, or malignancies such as KS. The presence of persistent fever, pancytopenia, liver dysfunction, and inflammatory markers raises clinical suspicion [27]. However, we lacked key diagnostic parameters such as ferritin, triglycerides, and soluble IL-2 receptor levels, and hemophagocytosis was not documented in the bone marrow biopsy which lowers the probability. These conditions may account for the patient’s rapid clinical decline despite appropriate antimicrobial therapy and are supported by rising inflammatory markers such as leukocytosis and elevated C-reactive protein levels.
Unfortunately, due to the patient’s transfer to another institution, we did not have access to clinical records from the final hospitalization, and no autopsy was performed. Thus, we were unable to confirm whether ART had been initiated, or whether there were additional clinical, laboratory, or imaging findings that might have supported a diagnosis of KS-IRIS or KICS. These limitations preclude definitive conclusions regarding the cause of death and highlight the diagnostic complexity in severely immunocompromised patients with overlapping opportunistic infections and malignancies.
Our case shares several features with the few reported instances of P. jirovecii involvement of the bone marrow in patients with advanced AIDS. As in previous reports, the patient exhibited profound immunosuppression, with a markedly low CD4 count and a high HIV viral load—both recognized risk factors for extrapulmonary dissemination. Notably, bone marrow involvement in our case was confirmed antemortem via bone marrow biopsy, whereas most previously documented cases were identified postmortem, often incidentally. This distinction reinforces the notion that disseminated P. jirovecii infection involving the bone marrow is likely underdiagnosed during life, particularly in the absence of targeted histopathologic evaluation.
4. Conclusions
P. jirovecii infection typically presents as pulmonary disease but may occasionally disseminate to extrapulmonary sites such as the bone marrow, particularly in patients with advanced immunosuppression. Bone marrow involvement is an uncommon but clinically significant finding that is usually identified through histopathologic evaluation and is associated with a high mortality.
This case contributes to the limited literature on disseminated P. jirovecii infection and highlights the importance of considering bone marrow involvement in people living with HIV who present with PCP and unexplained cytopenias. To date, no clinical trials have defined optimal treatment strategies for extrapulmonary or disseminated P. jirovecii infection, and it remains uncertain whether the standard 21-day cotrimoxazole regimen for pulmonary disease is adequate in these scenarios. Ongoing clinical awareness, histopathologic confirmation when feasible, and individualized clinical judgment remain essential in managing these rare and challenging presentations.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Alanio A. Bretagne S. Pneumocystis jirovecii detection in asymptomatic patients: What does its natural history tell us?F 1000 Research 2017673910.12688/f 1000 research.10619.128649366 PMC 5464226 · doi ↗ · pubmed ↗
- 2Tasaka S. Tokuda H. Pneumocystis jirovecii pneumonia in non-HIV-infected patients in the era of novel immunosuppressive therapies J. Infect. Chemother. Off. J. Jpn. Soc. Chemother.20121879380610.1007/s 10156-012-0453-022864454 · doi ↗ · pubmed ↗
- 3Telzak E.E. Cote R.J. Gold J.W.M. Campbell S.W. Armstrong D. Extrapulmonary Pneumocystis carinii Infections Clin. Infect. Dis.19901238038610.1093/clinids/12.3.3802193343 · doi ↗ · pubmed ↗
- 4Grier D.D. Lewis Z. Palavecino E.L. Bone marrow involvement by Pneumocystis jiroveci Br. J. Haematol.200914514910.1111/j.1365-2141.2008.07438.x 19533848 · doi ↗ · pubmed ↗
- 5Heyman M.R. Rasmussen P. Pneumocystis carinii Involvement of the Bone Marrow in Acquired Immunodeficiency Syndrome Am. J. Clin. Pathol.19878778078310.1093/ajcp/87.6.7803035916 · doi ↗ · pubmed ↗
- 6Rossi J.F. Pneumocystis carinii Infection of Bone Marrow in Patients With Malignant Lymphoma and Acquired Immunodeficiency Syndrome: Original Report of Three Cases Arch. Intern. Med.199015045010.1001/archinte.1990.003901401460302302020 · doi ↗ · pubmed ↗
- 7Rossi J.F. Pneumocystis carinii in Bone Marrow Ann. Intern. Med.198510286810.7326/0003-4819-102-6-868_13873199 · doi ↗ · pubmed ↗
- 8Unger P.D. Rosenblum M. Krown S.E. Disseminated Pneumocystis carinii infection in a patient with acquired immunodeficiency syndrome Hum. Pathol.19881911311610.1016/S 0046-8177(88)80327-73257197 · doi ↗ · pubmed ↗