Long-read plasmid sequencing strategy for evaluating intrinsic instability of tandem gene arrays
Hiroaki Takesue, Satoshi Okada, Takashi Ito

TL;DR
The paper introduces a sequencing method to study how unstable gene arrays are in yeast by measuring their length changes using long-read sequencing.
Contribution
A novel nanopore sequencing strategy is proposed to evaluate the intrinsic instability of tandem gene arrays in yeast.
Findings
Nanopore sequencing can directly determine the length distribution of gene arrays on plasmids.
The method allows assessment of array instability and identification of factors influencing stability.
Abstract
Tandem gene arrays are inherently unstable, particularly in recombination-prone organisms such as the budding yeast Saccharomyces cerevisiae . However, understanding the nature of this instability—how frequently and to what extent the target array contracts or expands within the genome—remains challenging. As a surrogate approach to this goal, we propose using nanopore long-read sequencing to directly determine the length distribution of a target gene array cloned onto a centromeric plasmid functioning as an artificial chromosome. This strategy will not only allow us to assess the intrinsic instability of the array but also help identify factors that may influence its stability.
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
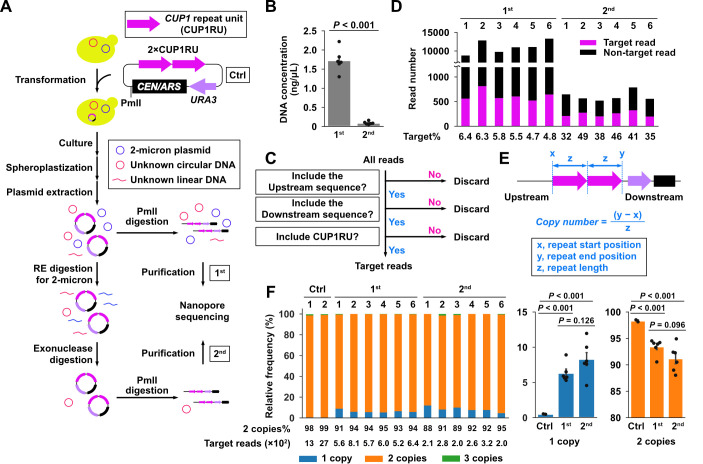 Figure 1
Figure 1|
Strain |
Genotype |
Available from |
|
YIT12685 |
|
Ito lab |
|
Plasmid |
Genotype |
Description |
|
34-29 |
YCpURA-2×CUP1RU |
A centromeric plasmid bearing two tandemly repeated copies of
|
- —Japan Science and Technology Agency (Japan)https://ror.org/00097mb19
- —Japan Society for the Promotion of Science (Japan)https://ror.org/00hhkn466
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsGene expression and cancer classification · Microfluidic and Bio-sensing Technologies · Single-cell and spatial transcriptomics
Description
Tandem gene duplication has played a central role in evolution, serving as a source of genetic diversity across various eukaryotic species, including pathogenic copy number variations in humans. It can lead to further recombination events between the repeat units, resulting in either expansion into a tandem gene array or contraction to a single-copy gene. Consequently, tandem gene arrays are inherently unstable, with the degree of instability varying among different arrays, presumably influenced by the nature of the repeat unit and the surrounding genomic environment.
To understand these factors, accurately assessing the frequency and extent of destabilization is crucial. While traditional Southern blot hybridization effectively determines the distribution of array lengths, it has limitations in detecting low-frequency variants. Long-read sequencing emerges as a promising alternative for this purpose. To maximize its potential at a reasonable cost, enriching target gene arrays through hybridization or Cas9-mediated excision is ideal; however, these approaches can be technically demanding.
Therefore, we propose a simple plasmid-based method as a surrogate assay for investigating the intrinsic instability of tandem gene arrays, leveraging cost-effective long-read sequencing. We used the budding yeast CUP1 array, which encodes a copper metallothionein, as our model system to evaluate instability.
We constructed the minimum CUP1 array, consisting of two 2-kb repeat units expected to be the most stable, on a centromeric plasmid marked with URA3 ( Figure 1A, YCpURA-2×CUP1RU). Cells transformed with YCpURA-2×CUP1RU were grown overnight in synthetic complete medium lacking uracil (SC−Ura) before plasmid extraction. We used two protocols to process the extracted plasmid DNA, which included both YCpURA-2×CUP1RU and the endogenous 2-micron plasmid. In the first protocol, the plasmid DNA was treated with the restriction enzyme PmlI, which cleaves YCpURA-2×CUP1RU but not the 2-micron plasmid ( Figure 1A, 1 ^st^ ), resulting in 1.73 ± 0.30 ng of purified DNA from half of an overnight 2-mL culture (~2.0 × 10 ^8^ cells) ( Figure 1B, 1 ^st^ ). In the second protocol, the plasmid DNA was treated with BsoBI and SphI to linearize the 2-micron plasmid without affecting YCpURA-2×CUP1RU, followed by exonuclease V (RecBCD) treatment to degrade the linear DNA. After heat inactivation of exonuclease V, PmlI digestion was performed to linearize YCpURA-2×CUP1RU ( Figure 1A, 2 ^nd^ ), resulting in 0.09 ± 0.04 ng of purified DNA from the other half of the culture ( Figure 1B, 2 ^nd^ ).
The 1 ^st^ and 2 ^nd^ samples purified from the yeast cells were subjected to Nanopore sequencing using the Native Barcoding Kit along with the Ctrl sample or the test plasmid prepared from E. coli for yeast transformation. The obtained reads were basecalled in super accuracy mode. Target reads containing both the vector and the CUP1 array were extracted from the obtained reads ( Figure 1C ). The 1 ^st^ and 2 ^nd^ samples contained the target reads at frequencies of 5.6% and 40.3%, respectively, demonstrating substantial enrichment of the target plasmid by eliminating the 2-micron plasmid and other linear DNAs ( Figure 1D ). The non-target reads in the 1 ^st^ sample were predominantly derived from ribosomal and mitochondrial DNAs, whereas those in the 2 ^nd^ sample exhibited no dominant DNA species. Given that the average length of non-target DNA fragments in the samples is similar to that of the target plasmid, we estimate the amounts of the target plasmid to be 0.096 ng (= 1.73 ng × 0.056) in the 1 ^st^ sample and 0.036 ng (= 0.09 ng × 0.403) in the 2 ^nd^ sample. These results suggest that the additional steps in the second protocol likely reduced the yield by 62.5% compared to the first protocol.
We then calculated the CUP1 copy number in the target reads using the formula shown in Figure 1E . The majority of reads contained two copies, while those with one copy (contraction) and three copies (expansion) accounted for an average of 7.32% and 0.36% of the reads, respectively ( Figure 1F ). Compared to the Ctrl sample obtained from E. coli , we observed a significant increase in the percentage of single-copy CUP1RU and a corresponding decrease in two-unit CUP1 arrays, regardless of the protocols used ( Figure 1F ). These results suggest that contraction occurs at a substantial frequency, even in the two-unit CUP1 array within yeast. Importantly, the difference in contraction frequencies between the 1 ^st^ and 2 ^nd^ samples was not statistically significant. Moreover, according to Oxford Nanopore Technologies' guidelines, maintaining most pores in an active sequencing state is crucial for preserving pore quality. Therefore, removing non-target DNA from extracted plasmid DNA samples is unnecessary as long as the number of samples is not excessively high.
The plasmid-based assay described above would provide a versatile method for identifying factors affecting tandem gene arrays, as it can reveal the distribution of array lengths. For the CUP1 array, it can be readily applied to examine various conditions, including the presence of copper in the medium and the absence of genes potentially affecting array stability. It would also facilitate the evaluation of the effects of the dCas9/nCas9-mediated interrogations we have developed for the targeted contraction and expansion of tandem gene array (Doi et al. 2021; Takesue et al. 2025).
Methods
Plasmid cloning
A centromeric plasmid containing two tandemly repeated copies of CUP1RU and the URA3 marker gene (YCpURA-2×CUP1RU) was constructed by seamless cloning using Golden Gate Assembly (New England Biolabs) and then transformed into E. coli using the Champion™ DH5α High competent cells (SMOBIO).
Yeast strains
The yeast strain used in this study was YIT12685 derived from BY4741 ( MAT a
- his3* Δ1
- leu2* Δ0
- met15* Δ0
- ura3* Δ0) (Brachmann et al. 1998). This study used standard culture media and genetic methods(Guthrie and Fink 1991). The yeast strain was transformed with the YCpURA-2×CUP1RU plasmid using the standard lithium acetate transformation protocol. Yeast cells were grown at 30°C overnight in 2 mL of the SC−Ura medium supplemented with 2% glucose.
Plasmid extraction
Yeast cells were washed with phosphate-buffered saline (PBS). After centrifugation, pellets were resuspended with spheroplast buffer [1 M sorbitol, 1× PBS pH 7.4, and 0.1 M EDTA] and 2.5 U Zymolyase (Zymo Research), and cell walls were digested at 30°C for 20 min, inverting every 5 min. After centrifugation for 3 min at 300× g , plasmid DNA was extracted from the spheroplast pellet using FavorPrep plasmid extraction mini kit (FAVORGEN). DNA concentration was calculated by Qubit 3.0 Fluorometer with Qubit dsDNA HS Assay System (Thermo Fisher Scientific). Non-target DNA was digested with restriction enzymes BsoBI (New England Biolabs), SphI-HF (New England Biolabs), and the exonuclease RecBCD (New England Biolabs), and the enzymes were denaturated at 80°C for 20 min. Plasmid DNA was linearized with the restriction enzyme PmlI (New England Biolabs) and purified with 0.4× AMPure XP (Beckman Coulter).
Nanopore sequencing
DNA libraries were prepared using the native barcoding kit SQK-NBD114 (Oxford Nanopore Technologies) according to the manufacturer’s instructions. The library was sequenced with the flowcell FLO-FLG114 R10.4.1 using the MinION sequencer (Oxford Nanopore Technologies). MinKNOW software was used to control the MinION device. The run time was set to 24 h. Base calling was performed using Dorado v0.7.3. The assessment of sequencing data was performed using NanoPlot (De Coster et al. 2018).
Copy number estimation from nanopore reads
We used nanopore sequencing data in FASTQ format and mapped reads to the S288c reference genome (version R64-2-1, http://sgd-archive.yeastgenome.org/sequence/S288C_reference/genome_releases/S288C_reference_genome_R64-2-1_20150113.tgz) using SAMtools (Danecek et al. 2021) and BEDtools (Quinlan et al. 2010). We then calculated normalized read count of each nucleotide using Bedgraph_norm_ratio.py (https://doi.org/10.5281/zenodo.11515696). Data were visualized with the IGV (Robinson et al. 2011). To eliminate the effect of read clipping and achieve a more accurate estimation of repeat unit number, we collected all reads containing the repeat unit using minialign (https://github.com/ocxtal/minialign). We then used its reference sequence as a query in a BLAST search (Altschul et al. 1990) against the collected reads and estimated the copy number based on the number of BLAST hits.
Generative AI and AI-assisted technologies
During the preparation of this work, the authors used GPT-4o to improve the readability of certain sentences. After using this tool/service, the authors reviewed and edited the content as needed and take full responsibility for the content of the publication.
Reagents
The yeast strain used in this study.
**: **
The plasmid used in this study.
**: **
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. 1990. Basic local alignment search tool. J. Mol. Biol. 215:403−410. https://doi.org/10.1016/s 0022-2836(05)80360-210.1016/S 0022-2836(05)80360-22231712 · doi ↗ · pubmed ↗
- 2Brachmann CB, Davies A, Cost GJ, Caputo E, Li J, Hieter P, Boeke JD. 1998. Designer deletion strains derived from Saccharomyces cerevisiae S 288C: a useful set of strains and plasmids for PCR-mediated gene disruption and other applications. Yeast 14:115−132. https://doi.org/10.1002/(sici)1097-0061(19980130)14:2%3C 115::aid-yea 204%3E 3.0.co;2-2 10.1002/(SICI)1097-0061(19980130)14:2<115::AID-YEA 204>3.0.CO;2-29483801 · doi ↗ · pubmed ↗
- 3Danecek P, Bonfield JK, Liddle J, Marshall J, Ohan V, Pollard MO, Whitwham A, Keane T, Mc Carthy SA, Davies RM, et al. 2021. Twelve years of SA Mtools and BC Ftools. Gigascience 10:giab 008. https://doi.org/10.1093/gigascience/giab 00810.1093/gigascience/giab 008PMC 793181933590861 · doi ↗ · pubmed ↗
- 4De Coster W, D'Hert S, Schultz DT, Cruts M, Van Broeckhoven C. 2018. Nano Pack: visualizing and processing long-read sequencing data. Bioinformatics 26:2666–2669. https://doi.org/10.1093/bioinformatics/bty 14910.1093/bioinformatics/bty 149PMC 606179429547981 · doi ↗ · pubmed ↗
- 5Doi G, Okada S, Yasukawa T, Sugiyama Y, Bala S, Miyazaki S, Kang D, Ito T. 2021. Catalytically inactive Cas 9 impairs DNA replication fork progression to induce focal genomic instability. Nucleic Acids Res. 49:954−968. https://doi.org/10.1093/nar/gkaa 124110.1093/nar/gkaa 1241 PMC 782627533398345 · doi ↗ · pubmed ↗
- 6Guthrie C, Fink GR. 1991. Guide to Yeast Genetics and Molecular Biology. Methods Enzymol. 194:1−863. Pub Med ID: 200578112005781 · pubmed ↗
- 7Quinlan AR, Hall IM. 2010. BED Tools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26: 841−842. https://doi.org/10.1093/bioinformatics/btq 03310.1093/bioinformatics/btq 033PMC 283282420110278 · doi ↗ · pubmed ↗
- 8Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP. 2011. Integrative genomics viewer. Nat. Biotechnol. 29:24−26. https://doi.org/10.1038/nbt.175410.1038/nbt.1754 PMC 334618221221095 · doi ↗ · pubmed ↗