A population description of CULAs using combined documentation of compound phenotypes according to the Oberg-Manske-Tonkin classification
Feikje Julia ten Cate, Margriet Harmke Maria van Doesburg

TL;DR
This study describes a population of patients with upper limb birth defects using a classification system that combines codes to capture complex cases.
Contribution
The study highlights the importance of combined documentation in the OMT classification for capturing compound phenotypes in congenital limb anomalies.
Findings
9.5% of patients required combined documentation for compound phenotypes.
Syndactyly, polydactyly, and camptodactyly were the most common diagnoses.
Recurring combinations included brachydactyly and clinodactyly, among others.
Abstract
Congenital upper limb anomalies (CULAs) exhibit a wide range of manifestations. For this reason, the Oberg-Manske-Tonkin (OMT) classification was introduced to achieve etiologically correct and universal classification. Combined use of codes has been advocated to prevent the loss of important phenotypic information in compound phenotypes. Therefore, the aim of our study was to present a description of our population over the last 10 years, using the most recent (2020) version of the OMT and combined documentation when necessary. All patients who visited our tertiary referral hospital between 2010 and 2020 were analyzed retrospectively and classified using the OMT. Combined registration was allowed in compound phenotypes and combinations were analyzed. Overall, 797 patients were included in our registry. Most anomalies were classified in the malformations group and 9.5% required…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
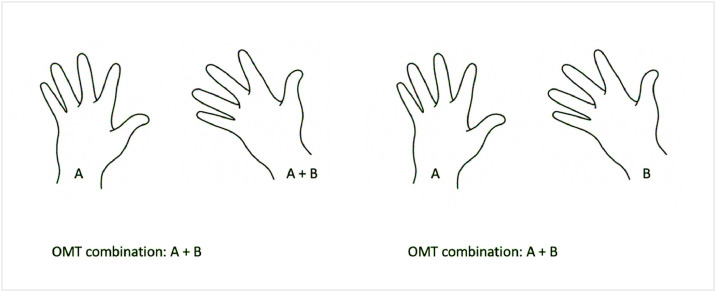 Figure 1
Figure 1Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsCongenital limb and hand anomalies · Genomic variations and chromosomal abnormalities · Genetic and rare skin diseases.
Introduction
Congenital upper limb anomalies (CULAs) are relatively uncommon, with an incidence of approximately 27 per 10,000 births.1 Given the wide spectrum of phenotypes displayed by these children, a uniform and comprehensive classification system is essential. The Oberg-Manske-Tonkin classification (OMT) is embraced by the International Federation of Societies for Surgery of the Hand (IFSSH) for the classification of CULAs because it allows for a more appropriate categorization of these anomalies, according to most recent scientific advances,2 than the previously used Swanson classification.3
Several epidemiologic studies have been conducted since its introduction. However, in 2018 Baas et al.4 noticed inconsistencies in combined documentation as most studies only registered the main anomaly per arm. Their study on the necessity of combined documentation found that a fifth of their patients required a combination of codes. Disregarding these compound phenotypes could lead to the loss of important information and hinder epidemiologic research and fair outcome comparison. Additionally, when compound phenotypes are well documented, subgroups within a population can be identified and could indicate an underlying etiology.
The purpose of our study was to provide a description of the CULA population at our tertiary referral hospital using the most recently modified (2020) version of the OMT classification,5 with combined documentation if required. Our second aim was to describe and analyze the code combinations registered in compound phenotypes.
Materials and Methods
This study adhered to the strengthening the reporting of observational studies in epidemiology (STROBE) guidelines for observational research
Sample
An observational cohort study was conducted. Data from all patients who visited our tertiary hospital for congenital hand or upper limb anomalies between April 2010 and April 2020 were reviewed retrospectively. Patients suitable for inclusion were all patients who visited the outpatient clinic for a first consultation or for follow-up. Patients with anomalies due to trauma, acquired anomalies, anomalies of the feet, and trigger fingers were excluded. Patient characteristics, medical history, location of the affected limb(s), and genetic test results were obtained. Radiographs and clinical photographs were acquired if they were available.
Classification strategy
All patients were reviewed by hand and classified by the first author using the 2020 update of the OMT classification.5 In the event of an ambiguity, a pediatric hand surgeon was consulted. Syndrome diagnosis was acquired from medical records, including genetic test results if available.
Multiple codes were registered in case of compound phenotypes. Bilaterally identical OMT diagnoses were counted and analyzed individually.
Combination analysis
The number of times a combination of two or more different codes was necessary was registered. In case of more than two different codes in one patient, all possible combinations were analyzed and counted. Additionally, information regarding the presence of a combination in the same extremity was documented.
Results
Registry characteristics
Between April 2010 and April 2020, a total of 797 patients were eligible for inclusion. Because patients could get more than one OMT classification code, a total of 874 OMT diagnoses were recorded. Overall, 428 OMT diagnoses were registered bilaterally and 446 unilaterally (left/right, 275/171; Table 1). Table 1 shows the number of times a certain code was seen unilaterally or bilaterally in a unique patient. Most anomalies were categorized in the malformations group. The first and second most commonly registered anomalies were ulnar and radial polydactyly, followed by cutaneous syndactyly and camptodactyly. In 84 patients (10.5% of the population), a syndrome was registered, with VACTERL association, Poland syndrome, and Greig Cephalopolysyndactyly being the most frequently observed (Table 2).Table 1. Distribution of CULAs according to the OMT classification, anomalies seen ≥25 times in bold font. Underlined numbers represent the total amount per main OMT groupTable 1OMT DiagnosisLeftRightBilateralTotalI. Malformations238156319713IA1i (Brachymelia)0101IA1iia (Symbrachydactyly spectrum-Poland syndrome)3104IA1iib (Symbrachydactyly spectrum-whole limb excluding Poland syndrome)1102IA1iiib (Transverse deficiency-segmental)95216IA2i (Radial longitudinal deficiency)65617IA2ii (Ulnar longitudinal deficiency)54211IA2iv (Radiohumeral synostosis)1001IA2v (Radioulnar synostosis)86519IA2vi (Congenital dislocation of radial head)5139IA2vii (Forearm hemiphyseal dysplasia, radial-Madelung deformity, or ulnar)563344IA4ia (Sprengel deformity)3205IB1i (Brachydactyly)731727IB1ii (Symbrachydactyly)2413441IB1iii (Transverse deficiency)87116IB1iv (Cleft hand-split hand foot malformation)42410IB2i (Radial longitudinal deficiency, hypoplastic thumb)991937IB2ii (Ulnar longitudinal deficiency, hypoplastic ulnar ray)4149IB2iii (Radial polydactyly)33371282IB2iv (Triphalangeal thumb)35412IB2iva (Triphalangeal thumb-five finger hand)3148IB2v (Ulnar dimelia-mirror hand)0202IB2vi (Ulnar polydactyly)5217123192IB4ia (Cutaneous -simple-syndactyly)2793470IB4iia (Osseous-complex-syndactyly)55515IB4iib (Clinodactyly)1042842IB4iid (Synostosis/symphalangism)0134IB4iiia (Syndromic syndactyly; e.g., Apert hand)0033IB4iiib (Synpolydactyly)3519IB4iiic (Unspecified axis-complex-not otherwise specified)0325II. Deformations94922IIA (Constriction ring sequence)94922III. Dysplasias2811100139IIIA2i (Macrodactyly)43411IIIA2ii (Aberrant flexor/extensor/intrinsic muscle)1001IIIB1i (Hemangioma)1001IIIB1ii (Vascular malformation)1001IIIB4ii (Enchondromatosis)2002IIIB4iii (Fibrous dysplasia)0011IIIB4vi (Other skeletal tumorous conditions)0011IIICia (Amyoplasia)102526IIICib (Distal arthrogryposis)112325IIICic (Arthrogryposis multiplex congenita-other)0088IIICiia (Camptodactyly)1572749IIICiib (Thumb in palm deformity)0099IIICiic (Isolated congenital contracture-other)2024Total:275171428874Table 2. Distribution of syndromic patients, represented in bold, are the top 3 most frequently documented syndromesTable 2SyndromeFrequencyA5. Bardet-Biedl5A6. Beals1A9. Catel-Manzke1A10. Cornelia de Lange1A12. Down5A14. Fancnoni pancytopenia1A15. Freeman Sheldon1A19. Greig cephalopolysyndactyly7A2. Apert3A21. Hemifacial microsomia (Goldenhar syndrome)1A22. Holt-Oram2A26. Leri-Weill dyschondrosteosis6A28. Moebius sequence1A31. Noonan1A32. Oculodentodigital dysplasia5A35. Pallister-Hall1A37. Pierre Robin1A38. Poland10A45. Split hand-foot malformation2A46. Thrombocytopenia absent radius3A47. Townes–Brock1A48. Trichorhinophalangeal1A49. Ulnar-mammary2A50. VACTERL association12B. Others: 16p11 deletion syndrome1B. Others: 22q11 deletion syndrome1B. Others: 4q-deletion syndrome1B. Others: Aarskog syndrome1B. Others: Branchio-oculo-facial syndrome1B. Others: Chitayat syndrome1B. Others: Ellis-van Creveld syndrome1B. Others: Frontomethafysair dysplasia1B. Others: Karsch–Neugebauer syndrome1B. Others: Vanishing white matter syndrome1Total84
Combinations
Among the 797 patients documented, 76 required more than one type of OMT code. Among those compound phenotypes, 65 required 2 different codes, 10 needed 3, and in 1 patient four different codes were registered (Table 3). In 17% (n = 13) of the patients with compound phenotypes, a syndrome was registered.Table 3. Number of patients with different OMT codesTable 3Total number of patients7971 OMT type7212 OMT types653 OMT types104 OMT types1
From these 76 patients who required multiple codes, 61 possible combinations of two different codes could be deducted. Thirty-four (38.2%) of those were extracted from phenotypes with threeor four different OMT codes, the rest were observed in patients with only two different codes. The original combinations observed in patients with three or four codes are shown separately in appendix A. Among the 61 possible different combinations, 16 were documented repeatedly and 45 only once.
In total, a combination of two different codes was counted 89 times. Twelve (13.5%) of these combinations of 2 OMT codes, were in opposite extremities. For example: Ulnar longitudinal deficiency (IA2ii) on the right hand and simple cutaneous syndactyly (IB4ia) on the left hand.
Sixteen recurring combinations accounted for 49.4% of all combined OMT diagnoses (n = 44).
To preserve comprehensibility, only the most frequently registered recurring combinations are shown in Table 4. The most commonly registered combinations were brachydactyly combined with clinodactyly, ulnar longitudinal deficiency of the upper limb combined with simple syndactyly, and simple syndactyly combined with clinodactyly. For the complete table of combinations see appendix A.Table 4. Most frequently observed recurring combinationsTable 4Combination of OMT DiagnosesFrequency (total)Unilaterally (i.e., same extremity)As part of larger combinationIB1i (Brachydactyly) and IB4iib (Clinodactyly)550IA2ii (Ulnar longitudinal deficiency entire upper limb) and IB4ia (Simple-cutaneous syndactyly)430IB4ia (Simple-cutaneous-syndactyly) and IB4iib (Clinodactyly)431IB4ia (Simple-cutaneous-syndactyly) and IB1i (Brachydactyly)331IB4iia (Osseous-complex-syndactyly) and IB4ia (Simple-cutaneous-syndactyly)320IB4ia (Simple-cutaneous-syndactyly) and IB2vi (Ulnar polydactyly)321IA2i (Radial longitudinal deficiency) and IA2vi (Congenital dislocation of radial head)320IB2i (Radial longitudinal deficiency, hypoplastic thumb) and IB4iia (Osseous-complex-syndactyly)330
Discussion
Similar to previous epidemiologic studies,4^,^6, 7, 8 syndactyly, polydactyly (radial and ulnar), and camptodactyly were most frequently observed. Unlike other registries, ours contained a relatively high count of Madelung deformities. This is probably the result of special expertise regarding that anomaly in our tertiary referral center.
Only 9.5% of our population required multiple OMT codes as opposed to more than 20.5% in the study by Baas et al.4. Their study also found a higher prevalence of triphalangeal thumbs combined with radial polydactyly. The latter could be the result of the high prevalence of a genetic mutation causing this anomaly in the southern Netherlands,9 near their hospital.
Regarding recurring combinations, brachydactyly and clinodactyly, ulnar longitudinal deficiency and simple syndactyly, and simple syndactyly and clinodactyly were the most frequently observed in our center. The high incidence of the last combination is likely to have a causal relationship. Untreated syndactyly could hinder the proper growth of one of the digits in a child and subsequently resulting in clinodactyly. All patients with this code combination, had it in the same digits that were involved in the syndactyly.
Strengths and limitations
The strength of our study lies in the use of the most recent version of the OMT and focus on combined documentation when necessary. Another strength of our study is that we provided a detailed description of the combinations observed.
Unlike previous studies, combinations of bilaterally identical OMT codes were analyzed individually to acquire a more accurate description of our population.
Baas et al.4 demonstrated the necessity of combined documentation, but did not specify whether the combination was observed in the same extremity or solely in the same patient. With this approach, valuable information on the type of combination and its subsequent phenotype may be lost. For example: patient 1 has a combination of anomaly A on the left hand and anomaly A and B on the right hand. In this case, the combination of A and B is documented (Figure 1). This results in a correct description of the patient. If patient 2 has a combination of anomaly A on the left and B on the right hand, this combination is counted similarly. However, this patient has a very different phenotype and therefore different prospects regarding for example surgical outcome. In 13.5% of the times that a combination was counted, it was observed in the opposite extremity. Our study demonstrates that disregarding the information on laterality when documenting combined diagnoses, could lead to loss of potentially important information. Therefore, we recommend specifying if a combination occurred in the same limb or digit when registering using the OMT.Figure 1. Ambiguous registration of OMT combinationFigure 1:
An important limitation of this study is that this study is a population description rather than a true epidemiologic analysis. Patients were seen in a tertiary referral center, therefore, no information on the prevalence or incidence can be provided. To provide these numbers, population data are required.
Another limitation is the retrospective design. Consequently, some classifications are based solely on the descriptions of physical examination by physicians, and several codes had no indication for an X-ray or medical photograph. Excluding these patients would have led to selection bias and an unreliable representation of our study population. Therefore, we decided to include patients regardless of the availability of these photographs.
In conclusion, we provided a detailed description of our population and thereby allow for comparison between facilities and provide information for research on underlying etiologies.
We agree with the proposition by Baas et al.4 to use combined documentation in the case of compound phenotypes and we additionally suggest including information on the specific location of the observed combination.
Conflict of interest
The authors declare no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Goldfarb C.A.Shaw N.Steffen J.A.The prevalence of congenital hand and upper extremity anomalies based upon the New York congenital malformations registry J Pediatr Orthop 372Mar 201714414810.1097/BPO.000000000000074827078227 PMC 5063649 · doi ↗ · pubmed ↗
- 2Oberg K.C.Feenstra J.M.Manske P.R.Developmental biology and classification of congenital anomalies of the hand and upper extremity J Hand Surg Am 3512 Dec 20102066207610.1016/j.jhsa.2010.09.03121134615 · doi ↗ · pubmed ↗
- 3Swanson A.B.A classification for congenital limb malformations J Hand Surg Am 11Jul 197682210.1016/s 0363-5023(76)80021-41021591 · doi ↗ · pubmed ↗
- 4Baas M.Zwanenburg P.R.Hovius S.E.R.Documenting combined congenital upper limb anomalies using the Oberg, Manske, and Tonkin classification: Implications for epidemiological research and outcome comparisons J Hand Surg Am 439Sep 201886910.1016/j.jhsa.2018.02.003e 1-869 e 1129573897 · doi ↗ · pubmed ↗
- 5Goldfarb C.A.Ezaki M.Wall L.B.The Oberg-Manske-Tonkin (OMT) classification of congenital upper extremities: Update for 2020 J Hand Surg Am 456Jun 202054254710.1016/j.jhsa.2020.01.00232093994 · doi ↗ · pubmed ↗
- 6Ekblom A.G.Laurell T.Arner M.Epidemiology of congenital upper limb anomalies in Stockholm, Sweden, 1997 to 2007: Application of the Oberg, Manske, and Tonkin classification J Hand Surg Am 392Feb 201423724810.1016/j.jhsa.2013.11.01424480684 · doi ↗ · pubmed ↗
- 7Goldfarb C.A.Wall L.B.Bohn D.C.Epidemiology of congenital upper limb anomalies in a Midwest United States population: An assessment using the Oberg, Manske, and Tonkin classification J Hand Surg Am 401Jan 201512713210.1016/j.jhsa.2014.10.038e 1-225534840 PMC 4276048 · doi ↗ · pubmed ↗
- 8Uzun H.Ozdemir F.D.M.Ustun G.G.Oberg-Manske-Tonkin classification of congenital upper extremity anomalies: The first report From Turkey Ann Plast Surg 853Sep 202024525010.1097/SAP.000000000000239732332389 · doi ↗ · pubmed ↗