FleQ finetunes the expression of a subset of BrlR-activated genes to enable antibiotic tolerance by Pseudomonas aeruginosa biofilms
Victoria I. Oladosu, Karin Sauer

TL;DR
The protein FleQ helps Pseudomonas aeruginosa biofilms resist antibiotics by working with SagS to control gene expression.
Contribution
FleQ's role in antibiotic tolerance is revealed, showing it works with SagS to regulate BrlR-activated genes.
Findings
FleQ and SagS work together to regulate BrlR-activated genes, contributing to antibiotic tolerance in biofilms.
Loss of FleQ or SagS leads to increased antibiotic susceptibility in P. aeruginosa biofilms.
FleQ interacts with SagS via the HisKA-Rec domain, suggesting a physical collaboration.
Abstract
The transcriptional regulator FleQ contributes to Pseudomonas aeruginosa biofilm formation by activating the expression and biosynthesis of matrix exopolysaccharides in a manner dependent on c-di-GMP. However, little is known about the role of FleQ in the antibiotic tolerance phenotype of P. aeruginosa biofilms. Inactivation of fleQ impaired biofilm formation and rendered biofilms susceptible to tobramycin and norfloxacin. The phenotypes were similar to biofilms inactivated in sagS encoding the orphan sensor SagS that promotes the switch from planktonic to biofilm growth via BfiSR and antibiotic tolerance via BrlR. While FleQ was found to contribute to biofilm formation independently of SagS and BfiSR, FleQ instead converged with SagS-dependent regulation at the level of BrlR. This was supported by multicopy expression of sagS failing to restore biofilm antibiotic tolerance by ΔfleQ to…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
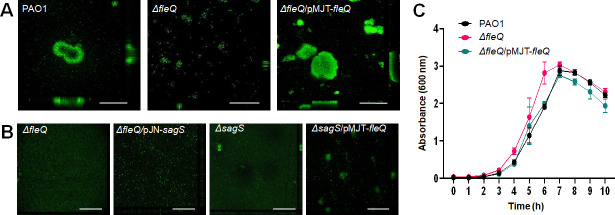 Fig 1
Fig 1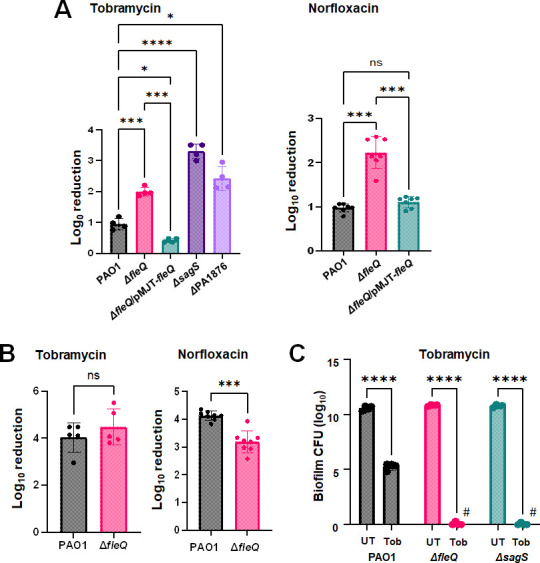 Fig 2
Fig 2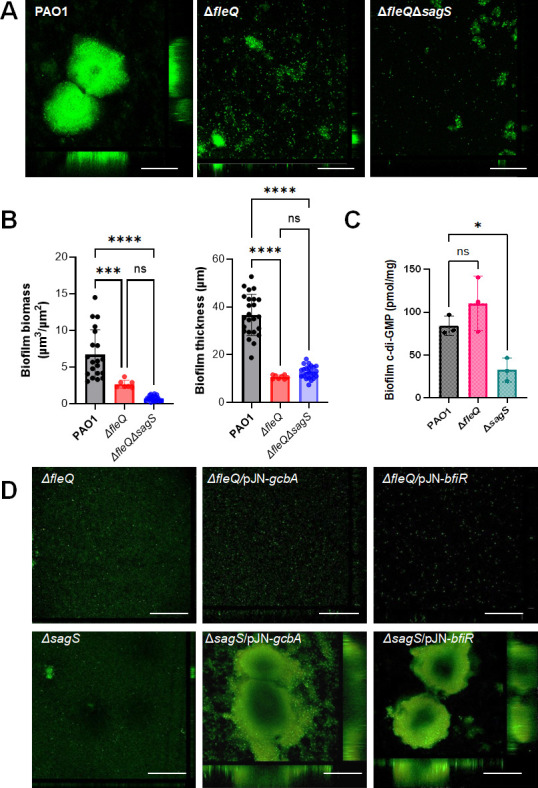 Fig 3
Fig 3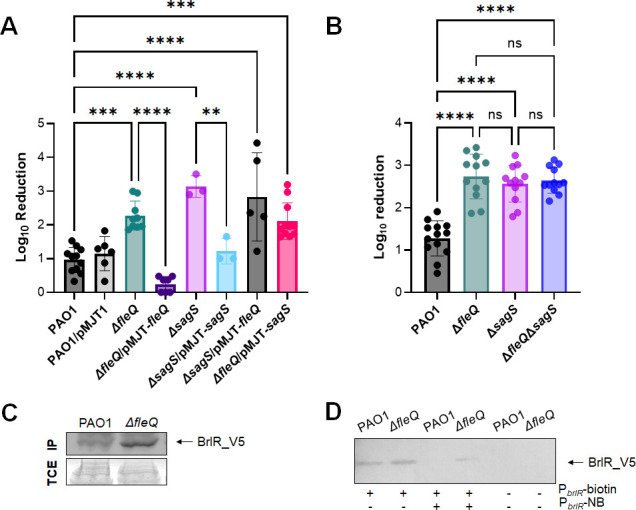 Fig 4
Fig 4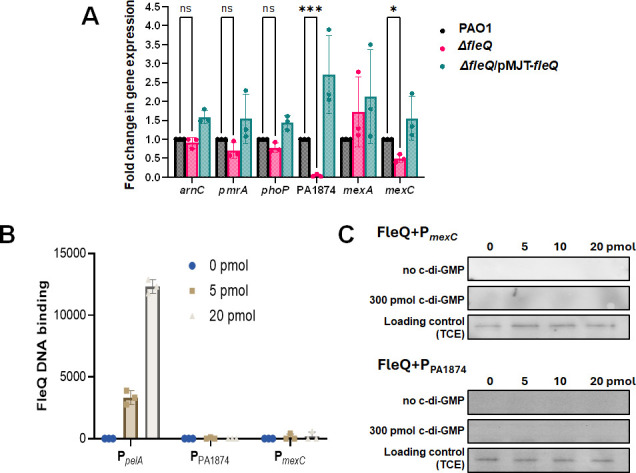 Fig 5
Fig 5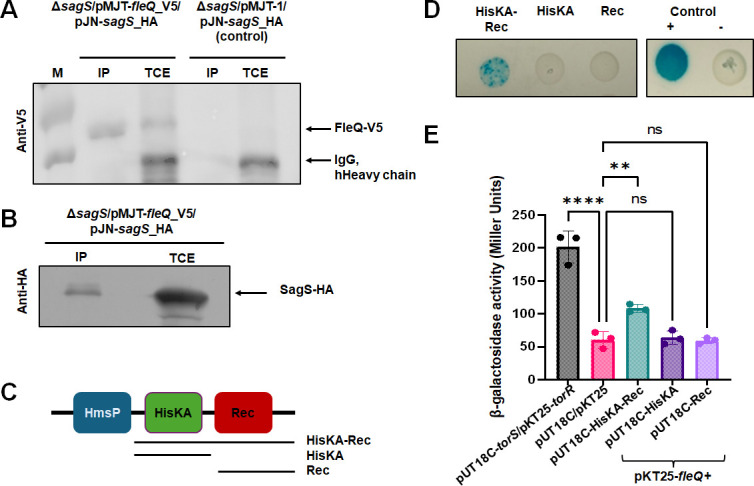 Fig 6
Fig 6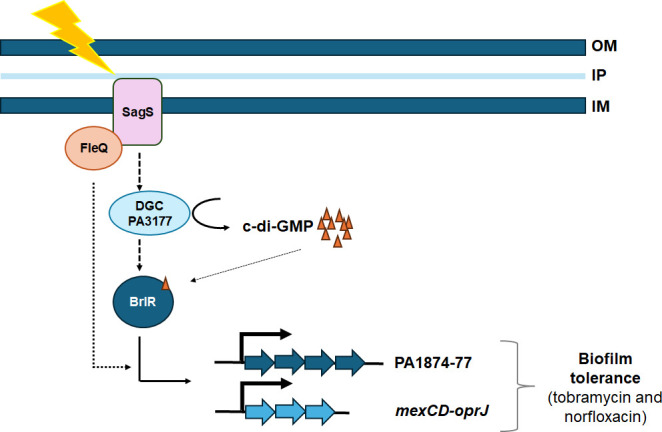 Fig 7
Fig 7| Strain | Total biomass (µm3/µm2) | Biofilm thickness (µm) |
|---|---|---|
| PAO1 | 6.48 (±3.14) | 9.21 (±3.42) |
| ∆ | 0.13 (±0.17) | 0.15 (±0.22) |
| ∆ | 5.87 (±3.64) | 6.34 (±3.95) |
| ∆ | 0.02 (±0.02) | 0.02 (±0.02) |
| ∆ | 0.02 (±0.02) | 0.01 (±0.02) |
| ∆ | 0.53 (±0.87) | 0.03 (±0.03) |
| ∆ | 0.38 (±0.38) | 0.31 (±0.35) |
| ∆ | 0.72 (±0.62) | 0.84 (±0.67) |
| Strains/plasmids | Relevant genotype description | Source |
|---|---|---|
| Strains | ||
|
| ||
| DH5α | F−, φ80 | Invitrogen corp. |
| DHM1 | F−, | ( |
|
| ||
| PAO1 | Wild-type strain PAO1 | B.H. Holloway |
| Δ | PAO1; Δ | ( |
| Δ | PAO1; Δ | ( |
| Δ | PAO1; Δ | This study |
| ΔPA1876 | PAO1, PA1876::IS | ( |
| Plasmids | ||
| pJN105 | Arabinose-inducible expression vector; pBRR-1 MCS; | ( |
| pJN- | C-terminal HA-tagged | ( |
| pMJT-1 | Arabinose-inducible expression vector; cassette of pJN105 cloned into pUCP18; | ( |
| pMJT-V5- | N-terminal V5-tagged | This study |
| pMJT- | C-terminally HA-tagged | ( |
| pJN- | C-terminally V5/6xHis-tagged | ( |
| pMJT- | pMJT-PA4843; | ( |
| pMJT- | C-terminally His6/V5-tagged | ( |
| pKT25 | Expression vector harboring the T25 fragment of CyaA; KmR | ( |
| pKT25- | pKT25 harboring TorR; KmR | ( |
| pKT25- | This study | |
| pUT18C | Expression vector harboring the T18 fragment of CyaA; AmpR | ( |
| pUT18C- | pUT18C harboring the Hpt domain of TorS; AmpR | ( |
| pUT18C-sagS_HisKA-Rec | pUT18C harboring SagS HisKA-Rec domain; AmpR | This study |
| pUT18C-sagS_HisKA | pUT18C harboring SagS HisKA domain; AmpR | This study |
| pUT18C-sagS_Rec | pUT18C harboring SagS Rec domain; AmpR | This study |
| CTX-P | pMini-CTX harboring C-terminal His6/V5-tagged | ( |
| pRK2013 | Helper plasmid for triparental mating; | ( |
| pEX18Gm | Allelic replacement suicide vector; pUC18 MCS; | ( |
| pEX- | Allelic replacement vector for | ( |
| Oligonucleotides | Sequences |
|---|---|
| Deletion | |
| sagSdel_for1_EcoRI | GCGCGCGCgaattcGCTGCAGGTAGCCGCTAC |
| sagSdel_rev1_BamHI | GCGCGCGCggatccCTCCGCATCGATGTTGTAGG |
| sagSdel_for2_BamHI | GCGCGCGCggatccGAGGAGCATCCCATCGACC |
| sagSdel_rev2_HindIII | GCGCGCGCaagcttGCCAGTCACGATGAGCGG |
| Cloning into pMJT-1 | |
| V5- | GCGCGCGCgctagcATG |
| | GCGCGCGCgagctcTCAATCATCCGACAGGTCGTC |
| Cloning into pKT25 | |
| linker-Xba1- | GCGCGCGCtctagaAATGTGGCGCGAACCAAACTC |
| linker-Sma1- | GCGCGCGCcccgggTCAATCATCCGACAGGTCGTC |
| Cloning into pUT18C | |
| sagS_HisKA-Rec-F | GCGCGCGCggatccGATGCTCGGGCGCATCTCGGTAG |
| SagS_HisKA-Rec-R | GCGCGCGCgaattcCTAGTCGCTCGCGGTGAGCGG |
| sagS_HisKA-F | GCGCGCGCggatccGATGCTCGGGCGCATCTCGGTAG |
| SagS_HisKA-R | GCGCGCGCgaattcCTAGCGCTGTTCTCCCGTGGTG |
| SagS_Rec-F | GCGCGCggatccGATGGTCAGCCCTCCGCTGCAG |
| SagS_Rec-R | GCGCGCGCgaattcCTAGTCGCTCGCGGTGAGCGG |
| Streptavidin bead DNA binding assay | |
| P_ |
|
| P- |
|
| P_PA1874_for |
|
| P_PA1874_rev |
|
| PbrlR-120F |
|
| PbrlR-120R |
|
| P_mexC_F |
|
| P_mexC_R |
|
| Sequencing primers | |
| pUT18C_MCS_For |
|
| pUT18C_MCS_Rev |
|
| pKT25_MCS_For |
|
| pKT25_MCS_Rev |
|
| sagS_deletion_check_rev |
|
| sagS_deletion_check_for |
|
| qRT-PCR primers | |
| |
|
| |
|
| |
|
| |
|
| |
|
| |
|
| |
|
| |
|
| |
|
| |
|
| PA1874-RT-F |
|
| PA1874-RT-R |
|
| |
|
| |
|
- —National Institute of Allergy and Infectious Diseaseshttp://dx.doi.org/10.13039/100000060
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsBacterial biofilms and quorum sensing · Bacterial Genetics and Biotechnology · Antibiotic Resistance in Bacteria
INTRODUCTION
Biofilms are characterized by bacteria growing in surface-associated communities or non-adherent aggregates of cells enclosed in a self-produced exopolysaccharide matrix (1). These sessile communities are characterized by heightened tolerance to antimicrobial agents and host immune response, with biofilms having been reported to be up to 1,000 times less susceptible to antimicrobial compounds when compared to their planktonic counterparts (2), and are the root cause for many chronic infections (3, 4). For example, biofilm formation by the opportunistic pathogen Pseudomonas aeruginosa is responsible for a substantial proportion of human nosocomial infections such as community-acquired pneumonia, burn wounds, chronic catheter-associated infections, foot ulcers, and corneal and mechanical ventilation-related infections (5–8), as well as chronic biofilm-associated infections within the lungs of people with cystic fibrosis (9).
In P. aeruginosa, the transition from the planktonic to the biofilm mode of growth coincides with the inverse regulation of genes encoding flagella and biofilm matrix components, including exopolysaccharide (Pel, Psl, alginate) and adhesins (e.g., CdrA) (10–14), as well as induction of antibiotic tolerance mechanisms (including efflux-pumps even when biofilms were grown in the absence of antibiotics) and increased levels of virulence determinants (15–17). The phenotypic changes noted upon transition to the sessile mode of growth are driven by multiple signaling systems such as the Rhl, Las, and PQS that are essential for the regulation of virulence and multi-cellular behaviors in response to quorum-sensing signaling molecules (18–20). Additionally, the transition to the multi-cellular mode of growth is regulated by the coordinated work of regulatory proteins interacting within large signaling networks such as LadS/RetS/GacAS/Rsm (Gac/Rsm), HptB, Wsp, and Pil-Chp systems that fine-tune alterations in the intracellular level of cyclic di-GMP (c-di-GMP) and, to a certain extent, levels of cyclic AMP (21–26). C-di-GMP is a ubiquitous bacterial second messenger that has emerged as a key regulator of biofilm formation, capable of regulating a myriad of cellular functions in bacteria including group behavior such as motility, surface attachment, and virulence (27–29). Additional regulators enabling the transition from the planktonic to the biofilm mode of growth include the two component systems SagS (25, 30, 31) and the transcriptional regulator FleQ (13, 14, 32). The sensor-regulator hybrid SagS has been linked to Gac/Rsm signaling and c-di-GMP regulation, with SagS facilitating biofilm development and the activation of biofilm-associated antimicrobial tolerance via two distinct pathways (reviewed in reference 25). Specifically, SagS promotes the switch from planktonic to biofilm growth via phospho-signaling to the BfiSR two-component regulatory system (TCS), which, in turn, contributes to the regulation of small regulatory RNA (sRNA) levels (30, 31). In addition, via a separate mechanism, SagS regulates the switch from an antimicrobial susceptible to a highly tolerant state via regulation of c-di-GMP levels and subsequent activation of BrlR, a c-di-GMP responsive transcriptional regulator of biofilm resistance (33–36). Specifically, SagS indirectly regulates brlR expression and BrlR function via the diguanylate cyclase PA3177 (35). The activation of PA3177 leads to the production of c-di-GMP, which subsequently binds to and activates BrlR (33–35).
The transcription factor FleQ contributes to biofilm formation by inversely regulating genes encoding flagella and exopolysaccharides in a manner dependent on the secondary messenger cyclic di-GMP (c-di-GMP) (14). Specifically, FleQ activates the expression of genes involved in biofilm formation when intracellular levels of c-di-GMP are high, while ceasing the expression of genes associated with the motile lifestyle, including flagellar gene expression (reviewed in 37). While the role of FleQ in the transition from planktonic to the biofilm mode of growth has been well established, little is known about the role of P. aeruginosa FleQ in the formation of antibiotic tolerant biofilms. A recent study indicated the biofilm matrix to reduce the penetration of positively charged antibiotics such as tobramycin, but not neutral antibiotics (38). While the matrix component responsible for sequestering tobramycin was not elucidated, tobramycin penetration was increased by the addition of cations in a dose-dependent manner, likely suggesting an involvement of charged matrix components such as Pel or eDNA, but not Psl. FleQ contributes to the biosynthesis of Pel and Psl matrix polysaccharides (13). However, it is not known whether FleQ contributes to the antibiotic susceptibility phenotype of biofilms. The goal of this study was to determine the role of FleQ in the formation of drug tolerant biofilms and determine whether FleQ intersects with other regulatory systems to enable the transition from the planktonic to the biofilm mode of growth and subsequent biofilm formation.
RESULTS
FleQ contributes to the P. aeruginosa biofilm architecture
FleQ is known to inversely regulate the expression of genes encoding flagella and exopolysaccharides in a manner dependent on the secondary messenger cyclic di-GMP (c-di-GMP) (13, 14, 32) and, thus, modulating motility and biofilm formation by Pseudomonas aeruginosa. To further explore the role of FleQ in the formation of antibiotic tolerant biofilms, we first assessed the ability of a P. aeruginosa mutant strain inactivated in fleQ to form biofilms under flowing conditions.
Biofilm formation was evaluated post 6 days of growth under continuous flow conditions. Visual analysis of the biofilm structure indicated ∆fleQ to form unstructured, flat surface-associated bacterial communities, while biofilms formed by wild-type PAO1 featured large microcolonies (Fig. 1A). COMSTAT analysis confirmed the stark difference in biofilm biomass accumulation and biofilm height (Table 1). Multicopy expression of fleQ restored biofilm formation by ∆fleQ to wild-type levels (Fig. 1A; Table 1). It is of interest to note that P. aeruginosa PAO1 and ∆fleQ demonstrated similar growth behavior under planktonic growth conditions (Fig. 1C), suggesting that the differences in the biofilm architecture are not due to differences in growth rates.
FleQ and SagS are both required for biofilm formation. Biofilms were grown in flow cells under flowing conditions for 6 days. Biofilms were stained using BacLight LIVE/DEAD prior to visualizing the biofilm architecture by confocal microscopy. (A) Representative confocal images of biofilms by P. aeruginosa PAO1, ΔfleQ and ΔfleQ/pMJT-fleQ. (B) Representative confocal images of ΔfleQ and ΔfleQ expressing sagS (ΔfleQ/pJN-sagS) and ΔsagS expressing fleQ (ΔsagS/pMJT-fleQ). White bar = 100 µm. All experiments were performed in triplicate. (C) Growth curves. P. aeruginosa PAO1, ΔfleQ, and ΔfleQ/pMJT-fleQ were grown in VBMM at 37°C and 220 rpm and the absorbance was determined at 600 nm. Experiments were carried out in triplicate. Error bars indicate standard deviations.
FleQ contributes to the antibiotic tolerance phenotype of P. aeruginosa biofilms
To assess the susceptibility of biofilms formed by ∆fleQ, surface-attached cells of wild-type and mutant strains were exposed to 150 µg/mL tobramycin for 1 h under flowing conditions. Biofilms formed by wild-type PAO1 cells demonstrated a less than 1 log reduction in viability following exposure to tobramycin, whereas ∆fleQ biofilm cells were reduced by >2.2 logs (Fig. 2A). Multicopy expression of fleQ restored the susceptibility phenotype of ∆fleQ biofilm cells to tobramycin to wild-type levels (Fig. 2A). It is of interest to note that little to no difference in the susceptibility to antibiotics was noted when grown planktonically (Fig. 2B).
*FleQ contributes to the susceptibility phenotype of P. aeruginosa biofilms. (A) Biofilms of indicated strains were grown in tube reactors under flowing conditions for 3 days and subsequently exposed to tobramycin (150 µg/mL) and norfloxacin (450 µg/mL) for 1 h under flowing conditions. Biofilm susceptibility was determined by log10 reduction. *, **, ***, ****, significantly different from the negative control (P < 0.05, P < 0.001, P < 0.0004, P < 0.0001, respectively) using ANOVA with Dunnett’s T3 multiple comparisons test. (B) P. aeruginosa PAO1 and ΔfleQ were grown planktonically to exponential phase and then exposed to tobramycin (50 µg/mL) and norfloxacin (50 µg/mL). Susceptibility was determined by log10reduction. ***, significantly different (P < 0.005) relative to PAO1, as determined using an unpaired t-test with Welch’s correction. ns, not significant. (C) Biofilm-MBC assays. P. aeruginosa PAO1, ΔfleQ, and ΔsagS were grown as biofilms for 3 days and subsequently treated for 24 h with tobramycin (300 µg/mL) under continuous flowing conditions before recovering and enumerating surviving cells. Biofilm susceptibility to tobramycin was determined by viable counts (biofilm CFU, obtained from biofilm tube reactors having an inner surface area of 25 cm2). #, no viable bacteria were detected. ***, significantly different from the untreated biofilm (P < 0.0001) using two-way ANOVA with Sidak’s multiple comparisons test. All experiments were carried out at least in triplicate. Error bars indicate standard deviations.
Previous findings indicated a protective role of biofilm matrix components by sequestering positively charged antibiotics such as tobramycin (and colistin) at the biofilm periphery (38). As FleQ contributes to the biofilm matrix by regulating the expression of pel and psl exopolysaccharides in biofilm (13, 14, 32, 39), we next asked whether the increased susceptibility of ∆fleQ biofilms was limited to the positively charged antibiotic tobramycin. We, therefore, evaluated the susceptibility phenotype of biofilms formed by ∆fleQ to the neutral fluoroquinolone, norfloxacin. Biofilms formed by ∆fleQ were more susceptible to norfloxacin compared to wild-type biofilms (Fig. 2A). Multicopy expression of fleQ restored the susceptibility phenotype of ∆fleQ biofilm cells to norfloxacin to wild-type levels (Fig. 2A), suggesting the increased susceptibility phenotype of biofilms formed by ∆fleQ to be independent of the abundance of exopolysaccharide.
Given the increased susceptibility phenotype of ∆fleQ biofilms, we, furthermore, determined whether FleQ affects the resistance of biofilm cells to killing, using a biofilm-MBC assay. P. aeruginosa wild-type and ΔfleQ cells were grown for 3 days as biofilms, after which time the medium was switched to the same medium containing 300 µg/mL tobramycin. This concentration has been previously demonstrated to eradicate ΔsagS but not wild-type biofilm cells (33). Therefore, biofilms by PAO1 and ΔsagS were used as controls. Following 24 h of exposure to the antibiotic under flowing conditions, the biofilms were harvested, and the surviving bacteria were enumerated. In agreement with previous findings (33), wild-type PAO1 biofilm cells were not eradicated, whereas no viable cells were recovered from the ΔsagS biofilms (Fig. 2C). Inactivation of fleQ affected recalcitrance to killing similarly to that of sagS, as no surviving cells were recovered from the ΔfleQ biofilms following the 24 h treatment with tobramycin (Fig. 2C).
FleQ does not contribute to biofilm formation in a SagS-dependent manner
The findings above suggested that FleQ contributes to the formation of antibiotic tolerant biofilms. The phenotype was somewhat reminiscent of biofilms formed by the P. aeruginosa ∆sagS ([Fig. 1A and 2A](#F1 F2)). sagS encodes the orphan sensor SagS that has previously been reported to independently promote biofilm development and the activation of biofilm-associated antimicrobial tolerance via the biofilm drug tolerance regulator BrlR (30, 33, 34, 40). BrlR is a global transcriptional regulator, regulating the expression of a multitude of targets including RND pumps and ABC transport systems, with inactivation of brlR coinciding with P. aeruginosa biofilms being rendered significantly more susceptible to 5 classes of antibiotics. The ABC transport system PA1774-77 has previously been reported to be regulated by BrlR and to contribute to the biofilm susceptibility phenotype to tobramycin as well as norfloxacin (41, 42).
Given the similarity in biofilm architecture and antibiotic susceptibility phenotype between ∆fleQ and ∆sagS biofilms, we next asked whether FleQ and SagS work synergistically or in concert to affect biofilm formation. We have previously demonstrated that multi-copy expression of certain genes, such as bfiR and brlR, encoding proteins that function downstream of SagS can restore biofilm phenotypes by ∆sagS to wild-type levels (30, 31). Thus, we reasoned that we could use similar epistasis assays to assess the functional relationship between SagS and FleQ. However, while multi-copy expression of fleQ restored biofilm formation by ∆fleQ to wild-type levels (Fig. 1A), multi-copy expression of sagS failed to rescue or enhance biofilm formation by ∆fleQ (Fig. 1B; Table 1). Likewise, multi-copy expression of fleQ in ∆sagS failed to rescue or enhance biofilm formation by the mutant strain (Fig. 1B; Table 1).
To discern whether FleQ and SagS to contribute to biofilm formation via distinct or convergent regulatory pathways, we also evaluated biofilm formation by the double mutant ∆fleQ∆sagS. Biofilms were grown for 3 days in 24-well microtiter plates, and biofilms by wild-type PAO1 and ∆fleQ were used as controls. While both ΔfleQ and the ΔfleQΔsagS double mutation formed biofilms with visibly reduced biofilm biomass than the parent strain PAO1, biofilms by ∆fleQ∆sagS appeared to be composed of even less biomass than ∆fleQ (Fig. 3A). COMSTAT analysis confirmed the overall reduction in biofilm biomass by ∆fleQ∆sagS relative to ∆fleQ; however, the difference was not significant (Fig. 3B). Nevertheless, these findings hinted at FleQ and SagS contributing to biofilm formation synergistically, via distinct, rather than convergent, regulatory pathways.
*FleQ contributes to biofilm formation in a manner independent of SagS and SagS downstream signaling. (A) Representative confocal images of 3-day-old biofilms formed by P. aeruginosa PAO1, ΔfleQ, and double mutant ΔfleQΔsagS. White bar = 100 µm. (B) Quantitative of the biofilm biomass and biofilm height by COMSTAT of biofilms formed by P. aeruginosa PAO1, ΔfleQ, and double mutant ΔfleQΔsagS. **, ***, significantly different (P = 0.0001, <0.0001, respectively) relative to PAO1 using ANOVA followed by the Bartlett’s test. Ns, not significant. (C) Total cellular c-di-GMP levels in 6-day-old biofilms formed by P. aeruginosa PAO1, ΔfleQ, and ΔsagS, as determined by HPLC quantitative analysis, followed by normalization relative to total cell protein content. , P < 0.05, relative to PAO1 using ANOVA followed by Dunnett’s T3 multiple comparisons test. (D) Representative confocal images of 6-day-old P. aeruginosa ΔfleQ and ΔsagS biofilms and mutant biofilms expressing bfiR and gcbA. gcbA encodes diguanylate cyclase GcbA, bfiR the two-component response regulator BfiR. White bar = 100 µm. All experiments were performed in triplicate. Error bars indicate standard deviations.
FleQ biofilm formation phenotypes cannot be rescued by the downstream targets of SagS
To further assess whether FleQ and SagS contribute to biofilm formation synergistically, we next asked if biofilms by the fleQ and sagS mutant strains differ in key characteristics such as c-di-GMP levels. FleQ affects biofilm formation via the regulation of biofilm matrix genes by P. aeruginosa, specifically the expression of genes contributing to the biosynthesis of Pel and Psl exopolysaccharides (13, 14). In contrast, SagS has been reported to contribute to biofilm formation by linking planktonic-specific Gac/Rsm-dependent signaling and the biofilm-specific TCS BfiSR to facilitate attachment-associated changes in the levels of the small regulatory RNA RsmZ (31, 43, 44) as well as c-di-GMP (30, 35), apparent by ∆sagS biofilm cells being characterized by significantly reduced c-di-GMP levels relative to PAO1 biofilm cells. In turn, biofilm formation by ∆sagS was restored to wild-type levels by multi-copy expression of gcbA encoding the diguanylate cyclase GcbA (30).
In agreement with previous findings (30, 45), ∆sagS biofilm cells were found to harbor significantly reduced c-di-GMP levels relative to PAO1 biofilm cells (Fig. 3C). In contrast, ∆fleQ biofilms were found to harbor higher levels of c-di-GMP (~100 pmol/mg c-di-GMP) than wild-type biofilms which were found to harbor on average 86 pmol c-di-GMP per mg cell protein (Fig. 3C). The difference in c-di-GMP levels was further supported by multicopy expression of gcbA encoding a diguanylate cyclase, restoring biofilm formation by ∆sagS to wild-type levels, without having an effect on the architecture of ∆fleQ biofilms (Fig. 3D; Table 1). Likewise, while multi-copy expression of bfiR encoding the cognate response regulator of the two-component regulatory system BfiSR (23, 31, 43), restored biofilm formation by ∆sagS to wild-type levels, expression of bfiR had no apparent effect on ∆fleQ biofilm formation (Fig. 3D; Table 1). The findings strongly suggest FleQ and SagS contribute to the formation of biofilms via distinct but cooperating mechanisms.
FleQ works in concert with SagS to enable the formation of antibiotic tolerant biofilms
As indicated above, the susceptibility phenotype of ∆fleQ biofilms in response to tobramycin and norfloxacin was similar to biofilms formed by P. aeruginosa mutant strains ∆sagS and ∆PA1876 (Fig. 2A), likely suggesting a role of both FleQ and SagS in the susceptibility phenotype of P. aeruginosa biofilms. To determine whether FleQ and SagS contribute to biofilm drug tolerance equally, we also made use of epistasis assays biofilm susceptibility assays in response to tobramycin. Multi-copy expression of sagS in ΔfleQ failed to restore biofilm susceptibility to tobramycin to wild-type levels, with ΔfleQ/pMJT-sagS biofilm cells demonstrating CFU reductions exceeding 2.5 logs following exposure to tobramycin for 1 hour (Fig. 4A). Likewise, overexpression of fleQ in ΔsagS cells did not rescue the biofilm susceptibility phenotype (Fig. 4A). Moreover, the double-mutant ∆fleQ∆sagS was significantly more susceptible to tobramycin compared to wild-type biofilms, but as susceptible as biofilms formed by the single mutants, ΔfleQ and ΔsagS (Fig. 4B), strongly suggesting FleQ and SagS to somewhat work in concert.
*FleQ is required for the tolerance of biofilm cells to tobramycin in parallel with SagS. (A, B) Susceptibility phenotype of biofilms grown in tube reactors under flowing conditions for 3 days to tobramycin (150 µg/mL). Susceptibility was determined by log10 reduction. **, ***, ***, significantly different from PAO1 biofilms (P < 0.005, P < 0.001, P < 0.0001) using one-way ANOVA, followed by Tukey’s multiple comparison test. ns, not significant. (C) Representative immunoblot showing FleQ-dependent abundance of BrlR. Total cell extracts (TCE) obtained from biofilms of indicated P. aeruginosa strains expressing a chromosomally located V5/His6-tagged BrlR under the control of its own promoter were probed for the presence of BrlR by immunoblot analysis (IB) using anti-V5 antibodies (anti-V5). The corresponding SDS-PAGE gel image obtained post-transfer indicates equal loading. Representative images are shown. (D) Representative immunoblot showing FleQ-dependent DNA binding capability of BrlR. BrlR-DNA binding was determined using streptavidin magnetic bead binding assays. Binding assays were carried out using 5 pmol of BrlR-V5/His6 protein obtained from the indicated strains and 1 pmol biotinylated PbrlR. Non-biotinylated PbrlR (PbrlR-NB) was used as specific competitor DNA in 20-fold excess. BrlR binding to PbrlR was detected by immunoblot analysis using anti-V5 antibodies. +/−, indicates presence/absence of specific probe or competitor. All experiments were performed in triplicate. Error bars indicate standard deviations
Inactivation of fleQ does not affect BrlR abundance or BrlR DNA binding
SagS has previously been reported to contribute to the switch from an antimicrobial susceptible to a highly tolerant state by indirectly activating the transcriptional regulator BrlR (30, 33, 34). BrlR, in turn, activates the expression of genes encoding multidrug efflux (MDR) pumps and ABC transporters, conferring tolerance to five classes of antibiotics including tobramycin and norfloxacin (17, 40). As ΔsagS biofilms have been characterized by the lack or significantly reduced abundance of active BrlR (30, 34), we first explored if the increased antibiotic susceptibility phenotype of biofilms formed by ΔfleQ may be due to decreased abundance of BrlR or alternatively, the presence of non-functional BrlR. Immunoblot analysis suggested the abundance of BrlR to be similar if not somewhat elevated in ∆fleQ biofilm cells relative to PAO1 (Fig. 4C). Moreover, using streptavidin pull-down assays and biotinylated PblrR (17, 36), BrlR produced by ∆fleQ biofilm cells was as capable of binding to the brlR promoter DNA and outcompeted by non-biotinylated competitor PbrlR DNA that abrogated the binding of BrlR, in a manner similar to BrlR obtained from wild-type biofilms (Fig. 4D).
FleQ finetunes the expression of a subset of BrlR-regulated genes
The above findings underscored that BrlR is both abundant and capable of DNA binding in ∆fleQ mutant cells. However, the question remained why biofilms formed by the ∆fleQ mutant strain, despite harboring functional BrlR at elevated abundance relative to wild-type biofilms, were susceptible to tobramycin and norfloxacin. We, therefore, asked if FleQ may somehow affect the functionality of BrlR, by assessing whether BrlR downstream targets are expressed, using quantitative reverse transcriptase PCR (qRT-PCR). Target genes included genes encoding Mex efflux pumps (mexA, mexC) (17), the ABC transporter PA1874-77 (41), and genes linked to colistin resistance, namely, pmrA, phoP, and arnC (46, 47). Inactivation or multicopy expression of fleQ had little to no effect on the BrlR target mexA, as well as genes linked to colistin resistance (pmrA, phoP, arnC) relative to wild-type biofilms (Fig. 5A). In contrast, however, significant differences in the transcript abundance were noted for the BrlR target PA1874, and somewhat for mexC in biofilms inactivated in or overexpressing fleQ relative to wild-type biofilms (Fig. 5A), indicating FleQ to finetune the expression of a subset of genes under regulatory control by BrlR previously linked to biofilm antibiotic tolerance.
*FleQ indirectly affects the expression of a subset of BrlR target genes. (A) Transcript abundance of BrlR-target genes in biofilms formed by ΔfleQ and complemented ΔfleQ/pMJT-fleQ mutant strains relative to the wild type (PAO1). Quantitative RT-PCR (qRT-PCR) was carried out using RNA extracted from 3-day biofilms grown in fivefold diluted VBMM. arnC encodes a component required for resistance to cationic antimicrobial peptide such as colistin; mexA and mexC encode components of multi-drug efflux pumps; PA1874 belongs to an operon that encodes ABC transport systems; phoP and pmrA encode two-component response regulators that control the expression of the arn operon. cysD was used as the housekeeping gene. Experiments were carried out in triplicate. Error bars indicate standard deviations. *, **, significantly different relative to the wild-type PAO1 (P < 0.5, P < 0.0001, respectively) using ordinary two-way ANOVA followed by Dunnett’s multiple comparison test. ns, not significant. (B, C) FleQ-DNA binding was determined using streptavidin magnetic bead binding assays, with FleQ binding to the respective promoter regions was detected by immunoblot analysis using anti-V5 antibodies, and subsequent analysis using ImageJ (48). Binding assays were carried out using 100 µg of total biofilm cell extract by PAO1 producing V5-tagged FleQ and 0–20 pmol biotinylated Ppel or PPA1874 or PmexC. (B) Quantitative analysis of FleQ DNA binding in the absence of additional c-di-GMP. (C) Representative immunoblots of FleQ DNA binding assays in the absence/presence of c-di-GMP and loading controls. Experiments were done in duplicate. Error bars indicate standard deviations.
To determine if FleQ directly affected the expression of mexC and PA1874, we determined whether FleQ is capable of binding to the promoter of the PA1874-77 and mexCD-oprJ operons. FleQ binding to the pel promoter was used as a control. While FleQ was capable of binding to the pel promoter (Fig. 5B) as previously reported (14), no binding to the promoter region of PA1874-77 and mexCD-oprJ was noted, neither in the absence nor in the presence of c-di-GMP (Fig. 5B and C), excluding a direct link between FleQ, PA1874-77, and mexCD-oprJ. This was further supported by the absence of a possible FleQ binding site in the promoter region of PA1874-77 and mexCD-oprJ (14, 49).
FleQ interacts with the orphan sensor SagS
The above findings indicated FleQ to not affect brlR expression or BrlR DNA binding, but instead to indirectly finetune the expression of the BrlR regulated genes PA1874-77 and mexCD-oprJ. However, this raised the question of how FleQ accomplishes this. As SagS has been described as a regulatory hub, connecting multiple overlapping signaling system that only indirectly modulates brlR expression to enable biofilm drug tolerance (30, 45, 50), we, therefore, asked whether FleQ interacts with SagS. To probe for interactions between FleQ and SagS, we made use of co-immunoprecipitation. Full-length FleQ was found to co-purify with SagS when HA-tagged SagS was used as bait proteins (Fig. 6A), while no FleQ was detected in the controls (Fig. 6A), confirming the specificity of the interaction between FleQ and SagS. This was further confirmed by the detection of SagS when V5-tagged FleQ was used as bait (Fig. 6B).
*FleQ interacts with SagS via its HisKA-Rec domain. (A) Representative image of immunoblot showing the abundance of FleQ in total cell extracts (TCE) and in pulldowns (IP) using SagS as prey. Strain ΔsagS/pMJT-1/pJN-sagS was used as control. M, protein ladder. (B) Representative image of immunoblot showing the abundance of SagS in total cell extracts (TCE) and in pulldowns (IP) using FleQ as prey. (C) Overview of SagS domains and SagS domain constructs. Lines underneath the domains indicate the composition of the SagS domain constructs, while the names of the resulting constructs are given next to the lines. (D) Representative images of bacterial adenylate cyclase two-hybrid assay (BACTH) results. DHM1/pUT18C-torS/pKT25-TorR was used as the positive control (+) and DHM1/pUT18C/pKT25 was used as the negative control (−). Bluish-green coloration represents a cleavage of X-gal by β-galactosidase indicative of protein-protein interaction. (E) Quantitative analysis of β-galactosidase activity using the Miller assay (51) of DHM1 strains co-expressing fleQ (pKT25-fleQ) and SagS-domain constructs (pUT18C -HisKA-Rec, pUT18C-HisKA, or pUT18C-HisKA-Rec). E. coli strains DHM1/pUT18C-torS/pKT25-torR and DHM1/pUT18C/pKT25 were used as controls. Experiments were done in triplicate. Error bars represent standard deviation. ** and ***, significantly different from the negative control (P < 0.01, P < 0.0001, respectively) using ANOVA followed by Dunnett’s T3 multiple comparisons test.
Previous findings indicated the switch function by SagS to be due to the modular composition of SagS (30) (Fig. 6C), with the cytoplasmic HisKA-Rec domains being required for its function (25, 30, 45). We, therefore, asked if FleQ interacts with SagS via one or both of its cytoplasmic domains, using bacterial adenylate cyclase‐based two‐hybrid (BACTH) assay in E. coli. Full-length FleQ and truncated SagS domain constructs, harboring only the HisKA-Rec, HisKA, or Rec domains of SagS (Fig. 6C), were fused to either the T18 or the T25 subunit of adenylate cyclase and then co-expressed in E. coli DHM1 strain to test for interaction. For controls, we made use of TorR and TorS (52), as well as a control strain harboring empty vectors (pKT25, pUT18C) (Fig. 6D), with the positive control being detected as blue colonies on medium containing X-Gal (5-bromo-4-chloro-3-indolyl-β-D-galactopyranoside) and the negative control coinciding with no color change (Fig. 6D). A positive interaction was noted between FleQ and the SagS construct harboring the HisKA and Rec domain (Fig. 6D). In contrast, little to no interactions were noticed between FleQ and the SagS constructs only harboring the HisKA or the Rec domain (Fig. 6D). Visual observations of protein-protein interactions were confirmed by Miller assays (Fig. 6E), suggesting FleQ to primarily interact with SagS via its HisKA-Rec domains.
DISCUSSION
The goal of this study was to explore the role of FleQ in the formation of drug tolerant biofilms and to determine whether FleQ intersects with other regulatory systems to enable the transition from the planktonic to the biofilm mode of growth.
We demonstrated that prolonged growth of ∆fleQ cells at the surface results in the formation of biofilms demonstrating severely reduced biomass and lack of complex three-dimensional architecture, including biofilm cell aggregates and microcolonies (Fig. 1). The resulting biofilms were highly susceptible to two antibiotics, the neutral norfloxacin and the positively charged tobramycin (Fig. 2A), with inactivation of fleQ disabling the recalcitrance of P. aeruginosa biofilms to tobramycin (Fig. 2C). Thus, while FleQ has been known to contribute to biofilm formation by expressing biofilm genes in a manner dependent on the levels of c-di-GMP (37), our findings expand on the role of FleQ from regulating the transition to the biofilm mode of growth to FleQ contributing to the antibiotic tolerance phenotype of biofilms. This was further supported by FleQ modulating the expression of mexC and PA1874 (Fig. 5A). Previous findings indicated that complete deletion of the genes encoding this pump, PA1874 to PA1877 (PA1874-1877), in both P. aeruginosa PA14 and PAO1 resulted in an increase in sensitivity to various antibiotics including tobramycin, gentamicin, norfloxacin, and ciprofloxacin, specifically when this mutant strain is growing as a biofilm (41, 42) (Fig. 2A). Moreover, inactivation of the respective pump has been shown to eliminate the recalcitrance of biofilms to killing by tobramycin (41, 42), in a manner similar to inactivation of fleQ (Fig. 2C).
PA1874-77 has previously been shown to be under the control of the transcriptional regulator BrlR (17, 40). However, we found no evidence of FleQ affecting BrlR abundance or BrlR-DNA binding (Fig. 4C and D), nor were we able to demonstrate FleQ directly regulating the expression of the PA1874-77 operon (nor mexCD-oprJ) (Fig. 5B). However, the expression of brlR, and in turn PA1874-77, has been shown to be dependent on SagS (30, 45). SagS and FleQ functioning synergistically to enable the formation of inherently antibiotic tolerant P. aeruginosa biofilms was apparent by epistasis assays (Fig. 4A), with multicopy expression of sagS failing to restore biofilm antibiotic tolerance by ΔfleQ to wild-type levels (and vice versa), and by biofilms formed by the ΔfleQΔsagS double mutant being as susceptible as ΔfleQ and ΔsagS biofilms (Fig. 4B). Given the synergy between FleQ and SagS and considering that SagS activates brlR while FleQ modulates the expression of BrlR target genes, the finding suggests FleQ contributes to biofilm tolerance by finetuning BrlR (upon activation in a SagS-dependent manner) (Fig. 7), apparent by FleQ only modulating a subset of BrlR downstream targets, specifically the PA1874-77 operon (Fig. 5A).
Model of FleQ finetuning BrlR target gene expression to modulate biofilm antibiotic tolerance. SagS indirectly contribute to the abundance and activation of the c-di-GMP responsive transcriptional regulator BrlR (36), likely by SagS contributing to the pool of available c-di-GMP generated by the diguanylate cyclase (DGC) PA3177 (33, 35). FleQ does not affect the abundance of BrlR or its DNA binding capability but indirectly affects the expression of a subset of BrlR target genes (17, 41). Dashed line, indirect effect of SagS on BrlR; dotted line, indirect effect of FleQ on BrlR, resulting in finetuning the expression of a subset of BrlR target genes.
While our findings failed to provide a direct link between FleQ and the regulation of mexCD-oprJ and PA1874-77, our findings indicate a close association between FleQ and SagS, with FleQ interacting with SagS via its HisKA-Rec domains (Fig. 6). The findings likely hint of the signaling network by the dual sensory switch protein SagS to comprise FleQ (or alternatively, the regulatory network of FleQ comprising SagS), with the two proteins affecting BrlR function, thus finetuning the BrlR regulon, to enable the formation of inherently tolerant P. aeruginosa biofilms (Fig. 7). However, the importance of the interaction between FleQ and SagS, and whether the two proteins need to interact for FleQ to finetune gene expression, is unknown and will be the focus of future studies.
MATERIALS AND METHODS
Bacterial strain, media, and growth conditions
P. aeruginosa PAO1 and indicated mutant strains used in this study are listed in Table 2. All planktonic cultures were grown in Lennox Broth (LB, BD Biosciences) in flasks at 220 rpm and 37°C. Biofilms were grown as indicated below. Antibiotics for plasmid maintenance were used at the following concentrations: 250 µg/mL carbenicillin (Carb) and 50–75 µg/mL gentamicin (Gm) for P. aeruginosa and 100 µg/mL ampicillin (Amp), 25–50 µg/mL kanamycin (Km), and 20 µg/mL gentamicin for Escherichia coli. Arabinose (0.1%) was added to induce the expression of genes cloned under the control of the arabinose inducible P_BAD_ promoter.
Strain construction
Isogenic mutants were constructed by allelic replacement using sucrose counterselection, as previously described (60), with the gene replacement vector pEX18Gm (59). Complementation and overexpression were accomplished by placing the respective genes under the control of an arabinose inducible promoter in the pJN105 or pMJT-1 vector (Table 2). Note that plasmid-borne fleQ harbored a V5-tag while sagS harbored an HA-tag (31). The V5-tag was introduced into fleQ via PCR amplification, using the primers and restriction sites indicated in Table 3. Plasmids were introduced via conjugation or electroporation. For bacterial two-hybrid assays, full length FleQ and SagS domain constructs (HiusKA, HisKA-Rec, Rec) were amplified using primers listed in Table 3 and cloned in frame into pKT25 and pUT18C plasmids. The generated recombinant plasmids (pKT25-fleQ, pUT18C-sagS, and pUT18C-HisKA-Rec) were subsequently introduced into E. coli DHMI strains by chemical transformation. All constructs were verified via DNA sequencing using primers listed in Table 3.
Planktonic growth curve
Overnight cultures of PAO1, ΔfleQ, and ΔfleQ/pMJT-V5-fleQ were inoculated as 1% inoculum into fresh VBMM with or without Carb (250 µg/mL). The optical density (OD) was measured every hour at 600 nm.
Biofilm growth
P. aeruginosa biofilms were grown in fivefold diluted VBMM medium using a continuous flow tube reactor system (1 m long size 13 or 14 silicone tubing, Masterflex, Cole Parmer, Inc.) at a flow rate of 0.1 mL/min or 0.2 mL/min, respectively, as well as in flow cells (BioSurface Technologies) at a flow rate of 0.2 mL/min (61–63). Following 6 days of growth, the biofilms were subjected to microscopy analysis or antimicrobial susceptibility testing as described below. For plasmid maintenance, gentamicin and carbenicillin were used in tube reactors at a concentration of 2 µg/mL.
Biofilm architecture analysis
Architecture of biofilms grown in flow cells was assessed via confocal laser scanning microscopy (CLSM). CSLM was carried out using a Leica TCS SP5 confocal microscope. Prior to confocal microscopy, biofilms were stained using the BacLight LIVE/DEAD viability stain (Life Technologies) at a 1/1,000 dilution in the growth medium, with 2 mL of the working BacLight LIVE/DEAD solution used per flow cell. The CLSM images were processed using LAS AF software v2.4.1. Quantitative analysis of the images was performed using COMSTAT MATLAB package and COMSTAT2 (64, 65).
Antibiotic susceptibility assays
All biofilms subjected to antimicrobial susceptibility assays were grown in tube reactors. Biofilms grown for 3 days were treated for 1 h with tobramycin (150 µg/mL) and norfloxacin (450 µg/mL). Following exposure of biofilms to the respective antimicrobial agents under flowing conditions, biofilms were harvested from tube reactors by squeezing the tubing, followed by the extrusion of the cell paste as previously described (63). To ensure complete disaggregation of cell aggregates, the resulting suspension was first homogenized for 15 s using a Biospec tissue tearer. The cell suspension was then serially diluted and spread plated onto LB agar. Viability was determined via CFU counts. Susceptibility is expressed as log_10_ reduction in viability.
Biofilm-MBC is defined as the concentration of antimicrobial where no additional killing is observed (17, 33, 40). We have previously shown that P. aeruginosa PAO1 biofilms demonstrate no significant increase in log reduction at concentrations greater than approximately 75 µg/mL of tobramycin after 24 h of treatment (17, 33, 40). Biofilms used for MBC assays were grown in continuous-flow tube reactors as described above for 3 days and subsequently exposed for 24 h to medium containing 300 µg/mL tobramycin. Following 24 h of treatment, biofilms were harvested, homogenized, and serially diluted. CFUs were determined via viable counts.
For planktonic susceptibility assays, overnight cultures of indicated P. aeruginosa strains were inoculated into fresh LB at 1% and grown for 3 h at 37ᵒC, at which point, the cells were treated for 30 min with tobramycin (50 µg/mL) or norfloxacin (50 µg/mL). Treated and untreated control cells were harvested by centrifugation and serially diluted and spread plated as described above.
Quantification of c-di-GMP levels
c-di-GMP was extracted in triplicate from 6-day biofilms of wild-type and mutant strains using heat and ethanol precipitation and quantitated essentially as described (66, 67). Briefly, c-di-GMP was extracted in triplicate using heat and ethanol precipitation followed. Supernatants were combined, dried using a Speed-Vac, and resuspended in 10 mM ammonium acetate. Samples (20 µL) were analyzed using an Agilent 1100 HPLC equipped with an autosampler, degasser, and detector set to 253 nm and separated using a reverse-phase C_18_ Targa column (2.1 × 40 mm; 5 µm) at a flow rate of 0.2 mL/min with the following gradient: 0–9 min, 1% B; 9–14 min, 15% B; 14–19 min, 25% B; 19–26 min, 90% B; 26–40 min, 1% B (buffer A, 10 mM ammonium acetate; buffer B, methanol plus 10 mM ammonium acetate). Commercially available cyclic di-GMP was used as a reference for the identification and quantification of cyclic di-GMP in cell extracts.
Immunoblot analysis and co-immunoprecipitation pull-downs
Co-immunoprecipitation pull-down assays were used to assess the interactions between SagS and FleQ in total protein cell extracts of cells co-producing V5-tagged FleQ and HA-tagged SagS. Pull-down assays were carried out using 2 mg of total cellular protein extracts. Following immunoprecipitation of V5-tagged proteins using immobilized anti-V5 antibodies at a concentration of 2 µg/mL, immunoprecipitation eluates were separated by SDS-PAGE and assessed by immunoblot analysis for the presence of HA-tagged prey SagS protein using anti-HA antibodies. Similarly, immunoprecipitation of HA-tagged protein was carried out using immobilized anti-HA antibodies, with the presence of V5-tagged proteins probed using anti-V5 antibodies. Densitometry of immunoblot bands was performed using the Gels Analysis tool of the ImageJ program (48).
Bacterial adenylate cyclase two-hybrid assays
Protein-protein interactions were assessed by the bacterial adenylate cyclase two-hybrid (BACTH) as previously described (68, 69). Proteins of interest were fused to the T18 or T25 fragment of Bordetella pertussis adenylate cyclase. The recombinant plasmids were co-transformed into E. coli DHM1 strain lacking the adenylate cyclase gene cya. Interaction of proteins of interest leads to heterodimerization of the T18 and T25 fragments, with interaction between the two hybrid proteins reconstituting the catalytic domain of adenylate cyclase, leading to cyclic AMP (cAMP) synthesis and transcription of the lac operon. DHM1 transformants were OD-adjusted to 0.1, and 2 µl per dilution spotted onto LB agar containing 50 µg/mL ampicillin, 50 µg/mL kanamycin, X-Gal (0.1 mM), and IPTG (isopropyl-β-D-thiogalactopyranoside, 0.1 mM). Plates were incubated at 30°C for up to 48 h, and colonies were examined for blue coloration. TorR and TorS protein served as positive controls (52), and the empty plasmids acted as the negative control. The interactions were quantitatively analyzed using β-galactosidase activity assays (51).
RNA isolation and qRT-PCR
Cells used for RNA isolation and qRT-PCR were grown as biofilms in fivefold diluted VBMM minimum media supplemented with carbenicillin (2 µg/mL) or gentamycin (2 µg/mL) in tube reactors for 3 days as described above. Biofilms were harvested into 3 mL RNAProtect, vortexed, and treated with 400 µg/mL lysozyme for 5 min prior to RNA isolation by the E.Z.N.A. Total RNA Kit I (OmegaBiotek). Prior to cDNA synthesis using the iScript cDNA synthesis kit (BioRad), RNA (1 µg) was DNAse treated using the TurboDNase-free (Invitrogen). qRT-PCR was carried out using BioRad CFX Connect Real-Time PCR Detection System and SsoAdvanced SYBR Green Supermix (BioRad), along with primers listed in Table 3. The housekeeping gene cysD was used. Relative transcript abundance was calculated using the CFX Manager Software (BioRad), by first normalizing the threshold cycle value (CT) to cysD, and then determining transcript abundance ratios. Expression of brlR downstream genes was determined using the 2^−ΔΔCT^method (70). Melting curve analyses were employed to verify specific single product amplification. The qRT-PCR analysis was performed using three biological triplicates.
Streptavidin magnetic bead DNA binding assay
DNA binding by BrlR and FleQ was assessed using the streptavidin magnetic bead DNA binding assay as previously described (60). Biotinylated target DNA fragments (PbrlR, PfleQ, PA1874*,* PmexC) were amplified using the primer pairs listed in Table 3. A total of 0–20 pmol of biotinylated target DNA was incubated for 30 min at room temperature with biofilm total cell extract containing 25 pmol of His_6_V5-tagged BrlR or 100 µg of V5-tagged FleQ (as indicated by immunoblot analysis using purified His_6_V5-tagged BrlR or biofilm total cell extract containing V5-tagged FleQ) in 25 mM Tris-Cl, pH 8, 5 mM MgCl_2_, 0.5 mM dithiothreitol, 1 mM EDTA, and 50 ng/ul poly(dI-dC). For specific competition, non-biotinylated PbrlR target DNA (0–50 pmol) was used. c-di-GMP (300 pmol) was added where indicated. Briefly, streptavidin magnetic beads (Thermo Scientific, 100 µg) were used to capture biotinylated DNA. Following three washes, the proteins co-purified with the biotinylated DNA were separated by 10% SDS/PAGE and assessed by immunoblot analysis for the presence of BrlR or FleQ using anti-V5 antibodies (Invitrogen). An aliquot prior to the addition of streptavidin magnetic beads was used to determine total BrlR or FleQ present in each DNA binding assay (loading control). Bands were visualized by chemiluminescence.
Statistical analysis
For pairwise comparison, a two-tailed Student’s t-test assuming equal variance or using single-factor analysis of variance (ANOVA) was used. Unless otherwise noted, all experiments were performed at least in triplicate using biological replicates.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Sauer K, Stoodley P, Goeres DM, Hall-Stoodley L, Burmølle M, Stewart PS, Bjarnsholt T. 2022. The biofilm life cycle: expanding the conceptual model of biofilm formation. Nat Rev Microbiol 20:608–620. doi:10.1038/s 41579-022-00767-035922483 PMC 9841534 · doi ↗ · pubmed ↗
- 2Davies D. 2003. Understanding biofilm resistance to antibacterial agents. Nat Rev Drug Discov 2:114–122. doi:10.1038/nrd 100812563302 · doi ↗ · pubmed ↗
- 3Costerton JW, Cheng KJ, Geesey GG, Ladd TI, Nickel JC, Dasgupta M, Marrie TJ. 1987. Bacterial biofilms in nature and disease. Annu Rev Microbiol 41:435–464. doi:10.1146/annurev.mi.41.100187.0022513318676 · doi ↗ · pubmed ↗
- 4Costerton JW, Stewart PS, Greenberg EP. 1999. Bacterial biofilms: a common cause of persistent infections. Science 284:1318–1322. doi:10.1126/science.284.5418.131810334980 · doi ↗ · pubmed ↗
- 5Ruiz M, Ewig S, Torres A, Arancibia F, Marco F, Mensa J, Sanchez M, Martinez JA. 1999. Severe community-acquired pneumonia. Risk factors and follow-up epidemiology. Am J Respir Crit Care Med 160:923–929. doi:10.1164/ajrccm.160.3.990110710471620 · doi ↗ · pubmed ↗
- 6Sadikot RT, Blackwell TS, Christman JW, Prince AS. 2005. Pathogen-host interactions in Pseudomonas aeruginosa pneumonia. Am J Respir Crit Care Med 171:1209–1223. doi:10.1164/rccm.200408-1044 SO 15695491 PMC 2718459 · doi ↗ · pubmed ↗
- 7Cole SJ, Records AR, Orr MW, Linden SB, Lee VT. 2014. Catheter-associated urinary tract infection by Pseudomonas aeruginosa is mediated by exopolysaccharide-independent biofilms. Infect Immun 82:2048–2058. doi:10.1128/IAI.01652-1424595142 PMC 3993445 · doi ↗ · pubmed ↗
- 8Restrepo MI, Babu BL, Reyes LF, Chalmers JD, Soni NJ, Sibila O, Faverio P, Cilloniz C, Rodriguez-Cintron W, Aliberti S, GLIMP. 2018. Burden and risk factors for Pseudomonas aeruginosa community-acquired pneumonia: a multinational point prevalence study of hospitalised patients. Eur Respir J 52:1701190. doi:10.1183/13993003.01190-201729976651 · doi ↗ · pubmed ↗