A Rare Case of Autoimmune Pulmonary Alveolar Proteinosis Developing During the Course of Eosinophilic Granulomatosis With Polyangiitis
Yoichi Dotake, Kentaro Tanaka, Shiro Fujisaki, Kenichi Shimobaba, Hirotoshi Kuroiwa, Midori Satomura, Hiromi Matsuyama, Koichi Takagi, Hideo Mitsuyama, Hiromasa Inoue

TL;DR
This paper reports the first case of autoimmune pulmonary alveolar proteinosis developing in a patient with eosinophilic granulomatosis with polyangiitis, highlighting the need for personalized treatment.
Contribution
This is the first reported case of autoimmune pulmonary alveolar proteinosis developing during eosinophilic granulomatosis with polyangiitis.
Findings
A 47-year-old woman with EGPA developed aPAP, confirmed by elevated anti-GM–CSF antibodies and imaging findings.
Mepolizumab successfully controlled EGPA relapse and allowed for steroid reduction, leading to improvement in aPAP.
The case suggests overlapping autoimmune conditions may require individualized immunomodulatory treatment and close monitoring.
Abstract
Eosinophilic granulomatosis with polyangiitis (EGPA) is an anti‐neutrophil cytoplasmic antibody (ANCA)‐associated vasculitis characterised by asthma, eosinophilia, and systemic inflammation, often involving the lungs. We present the case of a 47‐year‐old woman with EGPA who developed progressive ground‐glass opacities and a crazy‐paving pattern on chest computed tomography (CT). Bronchoalveolar lavage revealed milky fluid, and transbronchial lung biopsy showed periodic acid‐Schiff (PAS)‐positive eosinophilic granular material. Elevated anti‐granulocyte‐macrophage colony‐stimulating factor (GM–CSF) antibodies confirmed a diagnosis of autoimmune pulmonary alveolar proteinosis (aPAP). Corticosteroid tapering initially led to EGPA relapse, which was successfully controlled with mepolizumab, enabling further steroid reduction. Following this, the radiological findings of aPAP showed gradual…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
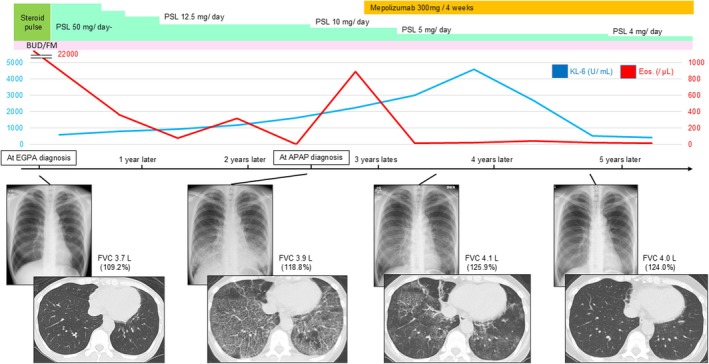 Figure 1
Figure 1 Figure 2
Figure 2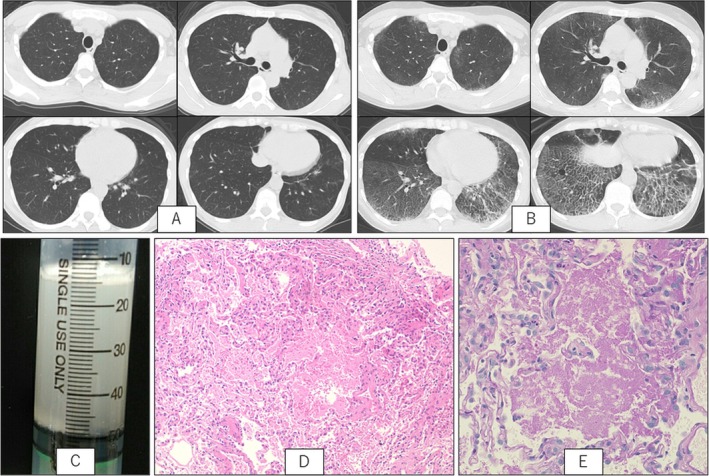 Figure 3
Figure 3Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsInterstitial Lung Diseases and Idiopathic Pulmonary Fibrosis · Vasculitis and related conditions · Eosinophilic Disorders and Syndromes