X‐Linked Sideroblastic Anaemia Caused by Intronic ALAS2 Variant Resulting in Highly Variable Expressive Phenotype in Male Siblings, a Case Report
James O'Connor, Niall Mannion, Caoimhe McKenna, Kerrie Sweeney, Aaron Niblock

TL;DR
Two brothers with X-linked sideroblastic anemia show very different symptoms due to an intronic mutation in the ALAS2 gene.
Contribution
Highlights variable expressivity of XLSA and challenges in diagnosing intronic ALAS2 mutations.
Findings
One brother required transfusions and a bone marrow transplant, while the other had mild symptoms.
An intronic ALAS2 variant was identified and reclassified as pathogenic after initial screening missed it.
Abstract
X‐linked sideroblastic anaemia (XLSA) is a rare hereditary disorder caused by mutations in the ALAS2 gene, essential for haem biosynthesis. We report two male siblings, the first of whom developed severe microcytic hypochromic anaemia requiring regular transfusions, iron chelation and an allogeneic bone marrow transplant, while his brother displayed only mild microcytic hypochromic indices without anaemia. Initial genetic screening did not identify a pathogenic variant. However, duo exome sequencing later revealed an intronic ALAS2 mutation, initially categorised as of uncertain significance and subsequently reclassified as pathogenic. This case underscores the diagnostic challenges posed by intronic mutations and the highly variable expressivity of XLSA, even among siblings. Trial Registration: The authors have confirmed clinical trial registration is not needed for this submission.
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
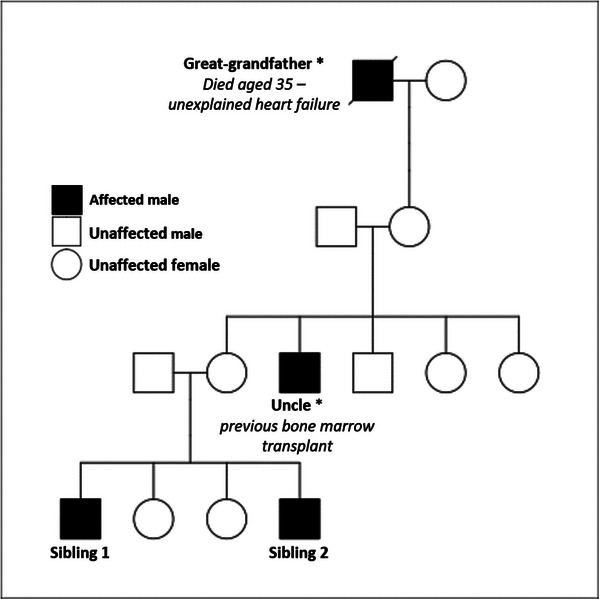 FIGURE 1
FIGURE 1Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsPorphyrin Metabolism and Disorders · Folate and B Vitamins Research · Heme Oxygenase-1 and Carbon Monoxide
Introduction
1
Sideroblastic anaemias are a heterogeneous group of inherited and acquired disorders characterised by the presence of ring sideroblasts in the bone marrow, reflecting defective haem biosynthesis [1]. Among inherited forms, X‐linked sideroblastic anaemia (XLSA) is the most common and results from mutations in the ALAS2 gene located on Xp11.21. This gene encodes 5‐aminolevulinate synthase, the enzyme catalysing the first step in haem biosynthesis [1]. Mutations in ALAS2 impair enzyme function, leading to reduced mitochondrial haem synthesis. The phenotypic expression of XLSA is highly variable, ranging from mild microcytosis to severe hypochromic microcytic anaemia with complications such as iron overload, ineffective erythropoiesis and splenomegaly [2]. Diagnosis relies on a combination of clinical findings, laboratory tests (e.g., blood counts, iron studies and peripheral smears), bone marrow biopsy with Prussian blue staining for ring sideroblasts and genetic testing [2].
We report two brothers with the same intronic ALAS2 mutation. Patient 1 presented with severe anaemia requiring blood product support and eventual haematopoietic stem cell transplantation, while Patient 2 exhibited only mild microcytosis. This case highlights the diagnostic challenges posed by intronic mutations and the variable clinical expressivity of XLSA.
Case Presentation
2
Patient 1
2.1
Patient 1, the younger sibling, first presented to his general practitioner (GP) at the age of 5 for an unrelated reason and was noted to have significant pallor. A full blood count revealed severe microcytic hypochromic anaemia. Initial management with repeated iron supplementation was ineffective. Over the next 5 years, his anaemia progressively worsened, necessitating regular blood transfusions and, as a result, iron chelation therapy. Other common causes of microcytic anaemias, including thalassaemia, were excluded.
A bone marrow biopsy demonstrated erythroblasts with nuclear‐cytoplasmic asynchrony, and Prussian blue staining revealed ring sideroblasts. These findings were consistent with inherited sideroblastic anaemia, but initial genetic testing failed to identify a causative mutation. Due to the severity of the condition and lack of response to pyridoxine, the patient eventually underwent an allogeneic bone marrow transplant.
Family pedigree analysis (Figure 1) identified an uncle who had received a bone marrow transplant and a great‐grandfather who had died of unexplained heart failure at the age of 35.
*Family pedigree demonstrating presumed X linked mode of inheritance. the great grandfather and uncle (for technical reasons) were not genetically proven to have the mutation.
Patient 2
2.2
Following the diagnosis of Patient 1, his older brother (Patient 2) underwent testing, which showed normal haemoglobin levels but low mean corpuscular volume (MCV) and mean corpuscular haemoglobin (MCH), along with elevated iron and ferritin. Although he exhibited no anaemia, these findings suggested a shared genetic basis. Due to the likely presence of the same underlying mutation, Patient 2 was excluded as a potential bone marrow donor.
Both brothers were found to carry the HFE C282Y variant, which may contribute to iron loading but did not fully account for the elevated ferritin levels in such young patients.
Genetic Investigations
2.3
Initial screening using a rare anaemia panel did not identify a pathogenic ALAS2 mutation. However, duo exome sequencing subsequently detected an intronic variant in the ALAS2 gene in both siblings. This mutation was initially classified as of uncertain significance but was later reclassified as pathogenic following the identification of additional, unrelated cases.
These findings confirmed a diagnosis of XLSA. Despite carrying the same mutation, the brothers exhibited highly variable clinical expressivity, with one being asymptomatic apart from mild microcytosis and the other requiring intensive treatment, including bone marrow transplantation.
Discussion
3
Sideroblastic anaemia is characterised by anaemia and the presence of ring sideroblasts in the bone marrow. Congenital sideroblastic anaemia (CSA) is a rare disease caused by mutations in genes involved in the haem biosynthesis. The most prevalent form of CSA is X‐linked sideroblastic anaemia, caused by mutations in the erythroid‐specific δ‐aminolevulinate synthase (ALAS2), which is the first enzyme of the haem biosynthesis pathway in erythroid cells [3]. The use of the next‐generation sequencing technology has recognised novel causative genes for CSA [4].
This case emphasises the diagnostic and clinical challenges associated with XLSA. The strikingly variable phenotypes seen in the two brothers underscore the unpredictable expressivity of ALAS2 mutations.
The phenotypic discordance observed in this case is consistent with prior reports of variability in expressivity of XLSA.
Cazzola et al. reported on two brothers with identical ALAS2 mutations whereby one brother developed severe anaemia and where the other remained largely asymptomatic [5].
This further supports the posit that even among individuals sharing the same variant of the XLSA mutation, clinical presentation is variable due to potential genetic, epigenetic or external factors.
Initial genetic screening, which focused on exonic regions of the ALAS2 gene, failed to detect the causative intronic mutation, highlighting a key limitation of current rare anaemia panels. Intronic mutations can impact splicing or gene regulation and should be considered when clinical suspicion of XLSA persists despite initial panel based screening. In this case, duo exome sequencing was crucial for identifying the pathogenic variant [6].
The identification of ring sideroblasts in the bone marrow remains a critical diagnostic hallmark, guiding further genetic investigation. This case also reinforces the importance of considering XLSA in patients with microcytic hypochromic anaemia unresponsive to iron supplementation and pyridoxine, particularly when there is evidence of iron overload at a young age.
Advances in genomic technologies such as exome sequencing are improving our ability to detect noncoding mutations, but these methods are not yet routinely available. This case demonstrates the need for expanded testing in patients with suggestive clinical and laboratory findings.
We also acknowledge that while variability is documented in XLSA, the precise contribution of other genetic or environmental factors remains unknown, and that additional undiscovered factors may contribute to the phenotypic differences observed in this case.
Author Contributions
A.N. and J.O'C. co‐wrote the manuscript, and with the assistance of N.M. gathered and analysed relevant clinical data. C.McK. reviewed and provided genetic data relevant to this case report. K.S. assisted with development of the family pedigree analysis. This article has been read and approved by all the authors.
Ethics Statement
The authors have nothing to report.
Consent
This work was published with consent from the patients.
Conflicts of Interest
The authors declare no conflicts of interest.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1G. Abu‐Zeinah and M. T. De Sancho , “Understanding Sideroblastic Anemia: An Overview of Genetics, Epidemiology, Pathophysiology and Current Therapeutic Options,” Journal of Blood Medicine 11 (2020): 305–318, 10.2147/JBM.S 232644.33061728 PMC 7524202 · doi ↗ · pubmed ↗
- 2J. Li , L. Chen , Y. Lin , and K. Ru , “Novel Mutations in the ALAS 2 Gene From Patients With X‐Linked Sideroblastic Anemia,” International Journal of Laboratory Hematology 42, no. 4 (2020): e 160–e 163, 10.1111/ijlh.13214.32297424 · doi ↗ · pubmed ↗
- 3T. Fujiwara and H. Harigae , “Molecular Pathophysiology and Genetic Mutations in Congenital Sideroblastic Anemia,” Free Radical Biology & Medicine 133 (2019): 179–185, 10.1016/j.freeradbiomed.2018.08.008.30098397 · doi ↗ · pubmed ↗
- 4S. Ducamp and M. D. Fleming , “The Molecular Genetics of Sideroblastic Anemia,” Blood 133, no. 1 (2019): 59–69, 10.1182/blood-2018-08-815951.30401706 PMC 6318428 · doi ↗ · pubmed ↗
- 5M. Cazzola , A. May , G. Bergamaschi , P. Cerani , S. Ferrillo , and D. F. Bishop , “Absent Phenotypic Expression of X‐Linked Sideroblastic Anemia in One of 2 Brothers With a Novel ALAS 2 Mutation,” Blood 100, no. 12 (2002): 4236–4238, 10.1182/blood-2002-03-0685.12393718 · doi ↗ · pubmed ↗
- 6D. R. Campagna , C. I. de Bie , K. Schmitz‐Abe , et al., “X‐Linked Sideroblastic Anemia due to ALAS 2 Intron 1 Enhancer Element GATA‐Binding Site Mutations,” American Journal of Hematology 89, no. 3 (2014): 315–319, 10.1002/ajh.23616.24166784 PMC 3943703 · doi ↗ · pubmed ↗