Multiple System Atrophy (Cerebellar Type) With Overlapping Progressive Muscular Atrophy Features and Genetic Erb-B2 Receptor Tyrosine Kinase 4 (ERBB4) Amyotrophic Lateral Sclerosis Variant: A Case Report
Enrique Lorenzo C Panganiban, Raymond L Rosales

TL;DR
A patient with Multiple System Atrophy and Progressive Muscular Atrophy features was found to have an ERBB4 gene mutation linked to ALS, highlighting overlapping neurodegenerative conditions.
Contribution
This case report presents a rare coexistence of MSA, PMA features, and an ERBB4 ALS variant, emphasizing the diagnostic complexity of overlapping neurodegenerative disorders.
Findings
The patient exhibited MSA-cerebellar type symptoms alongside lower motor neuron disease features resembling PMA.
Whole genome sequencing identified an ERBB4 gene mutation associated with an ALS variant.
The case highlights the coexistence of multiple neurodegenerative features in a single patient.
Abstract
Multiple system atrophy (MSA) is a progressive disease with Parkinsonism, dysautonomia, and cerebellar symptoms wherein patients can present with a broad range of confusing and overlapping findings attributable to various neuroanatomical substrates. Although possible, weakness is an unusual primary complaint, warranting further work-up for another neurodegenerative disease. The involvement of the more central structures, such as the locus coeruleus, pontine micturition center, and the cerebellum, can explain the wide range of symptoms. While Onuf's nucleus contributes to the urinary symptoms, anterior horn cells can implicate a motor neuron disease. Taking the varied neuroanatomical substrates into consideration, patients can present with a plethora of dysregulated motor symptoms. The authors share the course of a patient with clinically established MSA-cerebellar type and lower motor…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
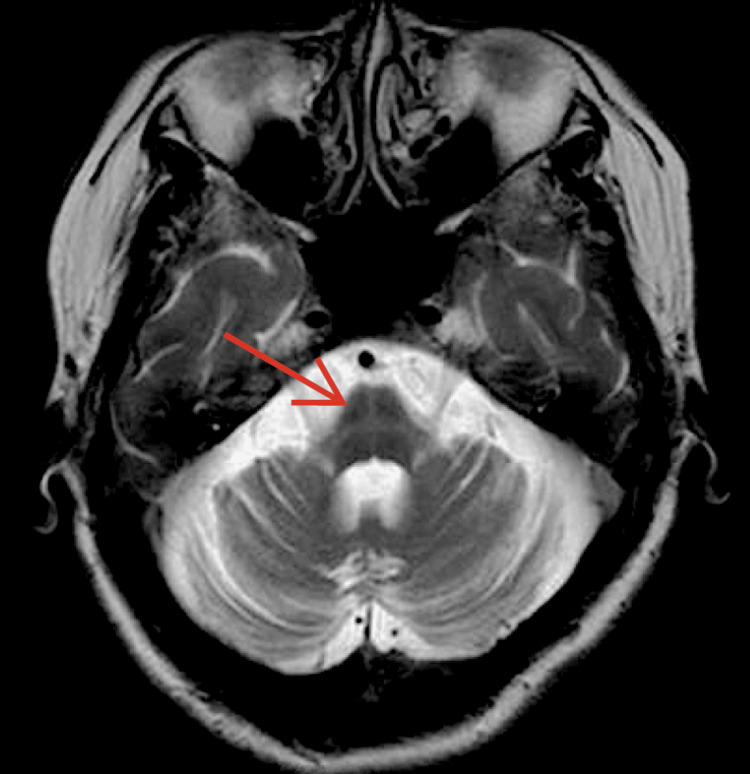 Figure 1
Figure 1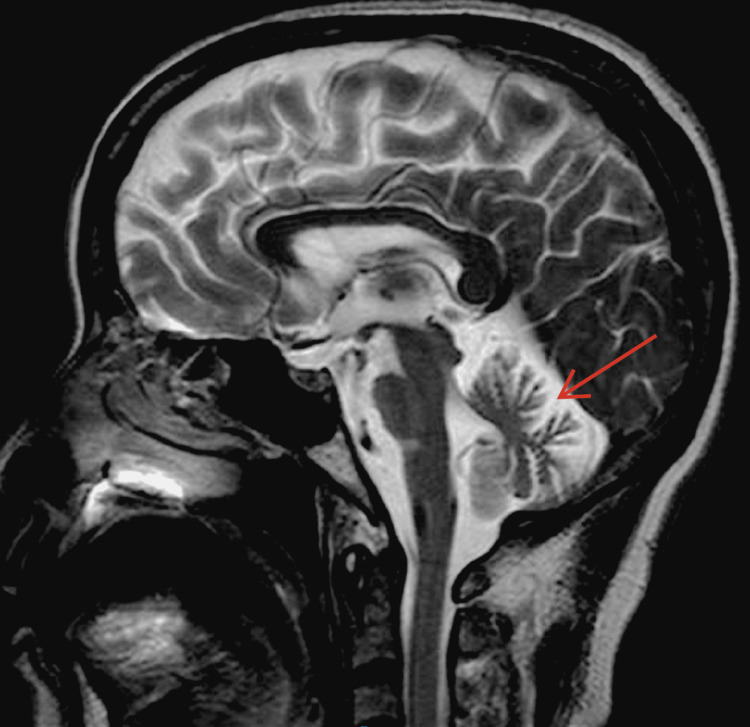 Figure 2
Figure 2| Muscles | Spontaneous activity | ||||
| Insertional activity | Fibrillations | Fasciculations | PSW | Motor unit potentials | |
| Right tibialis anterior | Normal | - | - | - | 6 Hz tremor, reduced recruitment pattern |
| Left tibialis anterior | Increased | - | - | ++ | Long duration, reduced recruitment pattern |
| Right triceps | Increased | - | - | + | Long duration, reduced recruitment pattern |
| Right first dorsal interosseous | Increased with CRD | + | + | ++ | Long duration, reduced recruitment pattern |
| Genioglossus | Increased | - | + | - | Long duration, reduced recruitment pattern |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsAmyotrophic Lateral Sclerosis Research · Parkinson's Disease Mechanisms and Treatments · Neurological diseases and metabolism
Introduction
As an alpha-synucleinopathy, characterized by the presence of glial cytoplasmic inclusions, multiple systemic atrophy (MSA) is an adult-onset, sporadic, relentlessly progressive neurodegenerative disease characterized by varying degrees of Parkinsonian symptoms, dysautonomia, and cerebellar involvement. It is caused by selective atrophy and neuronal loss of the striatonigral and olivopontocerebellar systems, giving rise to its MSA-Parkinsonian (MSA-P) and MSA-cerebellar (MSA-C) subtypes [1,2]. Though the presenting hallmark of patients with MSA is recurrent syncope, as a result of orthostatic hypotension, a percentage of them may present with urinary incontinence as an initial complaint. However, there have been reports that MSA patients can also present with polyminimyclonus, rapid progression to wheelchair confinement, cognitive impairment, hallucinations, and even pathological laughter or crying [1,3].
On the other hand, while weakness can present in patients with Parkinsonism, it is not a common initial manifestation. There could be a possibility of an overlapping motor neuron pathology. Although differentiating between upper motor neuron versus lower motor neuron diseases can aid in the diagnosis, familial and sporadic types of MNDs can further narrow the differentials. Interestingly, the spine is involved in patients with MSA since the Onuf's nucleus, located in the sacral region, lies close to the somatic motor neurons within the anterior horn of the spinal cord, which can explain some symptoms of motor neuron diseases [4].
Given the multiple presentations, intracranial imaging and neurophysiological studies can confirm the diagnosis in such cases. However, the signs and symptoms can still be explained by certain neuroanatomic substrates such as the locus coeruleus. Genetic predisposition plays a significant role in symptom development, and prudent investigation of these defects should always be offered to the patient. Hence, the authors share how multiple neurodegenerative movement disorders can coexist in a single patient, specifically, clinically established MSA-C and lower motor neuron disease findings at par with progressive muscular atrophy (PMA), but tested positive for an ERBB4 gene, a heterozygous variant of uncertain significance, associated with the autosomal dominant amyotrophic lateral sclerosis (ALS).
Case presentation
The case presented is a 65-year-old Chinese individual, ambulatory, with no history of trauma, but experienced bilateral gradual lower extremity weakness and gait imbalance, without any sensory deficits, which she had attributed to her low back pain from her previous spinal surgery five years prior to admission. She was able to perform her activities of daily living, but with increasing effort to lift her legs and developing slowness in gait. Three months later, she was admitted to a different hospital for urinary tract infection (UTI), urinary incontinence, and neurologic assessment. At the time, she presented with bilateral leg weakness, without sensory deficits, and an intact anal sphincteric reflex. A faint vertical, linear T2 hyperintensity within the pons, and cerebro-cerebellar atrophy was seen on non-contrast enhanced cranial MRI, while the first set of neurophysiological studies for her leg weakness yielded inconclusive results. She minimally improved on carbidopa-levodopa and did not return to her baseline.
The interim showed progressive weakness and more frequent urinary incontinence. Follow-up diagnostics showed a more distinct pontine linear T2 hyperintensity and further cerebellar atrophy on another MRI scan. During the pandemic, she was lost to follow-up and lived alone. She gradually deteriorated, requiring a walker and a diaper. Her voice became weak, poorly modulated, and staccato-like. She became irritable, bored, and disinterested in her family, which they had thought was due to the COVID lockdown.
Two years prior to admission, she was admitted for an episode of fall. A "hot-cross bun" sign within the pons was definitively seen on cranial MRI, and medications no longer improved her condition (Figures 1, 2). Aside from her recurrent UTI, she constantly experienced pre-syncopal episodes with orthostatic hypotension and became wheelchair-bound with asymmetric limb tremors. She was eventually managed as a case of MSA-C with features of PMA and was able to ambulate with assistance again after rehabilitation therapy. However, over the next year, she worsened again, now bed-bound with increasing memory lapses, misplaced laughing fits during conversations, beginning rapid eye movement (REM) sleep changes, described as acting out her dreams instead of the usual atonic state of sleep, complex visual hallucinations, daily pre-syncopal episodes during transfers, uncontrollable involuntary small amplitude, repetitive, non-rhythmic twitching of the hands and feet while awake and asleep, lasting for a few seconds, and gradual arm weakness. She was readmitted for treatment.
Cruciform pontine hyperintensity ("hot-cross bun" sign) seen in patients with MSA on cranial MRI (axial view)MSA - multiple system atrophy; MRI - magnetic resonance imaging
Marked cerebellar atrophy on cranial MRI (sagittal view)
Physical examination elicited emotional lability, bilateral horizontal gaze nystagmus, saccadic dysmetria, ataxic dysarthria, intention and resting tremors, rigid arms, proximal muscle weakness, atrophy of the left gastrocnemius more than its contralateral counterpart, polyminimyoclonus, tongue fasciculations, areflexia, and hand dystonia. A repeat of neurophysiological testing (Table 1) revealed findings for a PMA. The whole exome sequencing yielded an ERBB4 gene mutation (p. Arg979Gln), which was classified as a heterozygous variant of uncertain significance associated with the autosomal dominant form of ALS-19. However, family history had denied any relative with similar symptoms. For two weeks, she was treated with fudrocortisone (100 mcg per tablet) once daily, improving her orthostatic hypotension, which was not documented by the conventional definition, and daily coenzyme Q10 tablets and intravenous edaravone, barely aiding her weakness. Furthermore, she was treated again for her urinary tract infection and underwent rehabilitation therapy twice a day, thus improving her leg strength, but she was still discharged wheelchair-bound. Two years afterward, she had succumbed to systemic complications.
Discussion
The case exemplifies how movement disorders can overlap with and even mask diseases of the motor neurons. The documented cerebellar symptoms were clearly seen in the progressive atrophy of this structure. However, the underlying neuroanatomic substrates attributed to blood pressure and urinary incontinence can be localized to numerous supraspinal structures involving the central autonomic network, such as the locus coeruleus within the pons. This provides much of the brain's neuromodulator, norepinephrine, which contributes to wakefulness, behavioral flexibility, memory, and cognition [5]. Historically, the "hot-cross bun" sign or the cruciform pontine hyperintensity was thought to be a pathognomonic sign specific for MSA. Although it does implicate structures that contributed to the patient's gradual deterioration and multiple symptoms, it has long since been refuted that this is the only pathology it is linked to and can be seen in other conditions [6]. Nonetheless, this radiologic finding increased the index of suspicion for MSA-C as the culprit of the patient's presentation.
However, the central location of these structures will not fully explain the lower motor neuron findings, such as the proximal asymmetric weakness, atrophy, and areflexia, warranting the search for another underlying pathology, which was a possible PMA despite the inconclusive findings on the first set of neurophysiological studies. Also essential to urinary bladder control is the sacral region of the spinal cord, where Onuf's nucleus is located. This structure may be involved in MSA; however, it is the adjacent motor neurons within the anterior horn of the lumbosacral spinal cord that can justify the weakness [4]. Given that the patient presented with Parkinsonian symptoms, dementia-like symptoms, and ALS, a corticostriatospinal degeneration (Muro disease) was considered, like the Japanese studied in the Kii-peninsula, but was no longer considered given the prominent dysautonomia in the case [7]. Despite the limited effective management in preventing both MSA and ALS, symptomatic treatment still offers some clinical response. In theory, supplying the decreased coenzyme Q10 levels should improve the mitochondrial dysfunction and vulnerability to oxidative stress in patients with MSA [8]. While edaravone attempts to increase mitochondrial membrane potential, and ATP levels, and reduce reactive oxygen species [9-10].
Though PMA was considered, it was an ALS variant involving the ERBB4 gene that the results indicated toward. The ErbB signaling pathway plays a key role in the development, maintenance, and repair of motor neurons within both the central and peripheral nervous systems. Part of the epidermal growth factor family of extracellular ligands, neuregulins bind to the ErbB family of receptor tyrosine kinases, whose downstream effects contribute to diverse neuronal function such as cell differentiation, recovery after nerve injury, synaptic and neuromuscular junction development, maturation and myelination of Schwann cells, and regulation of neurotransmitter release, especially the ErbB4 receptor family [11]. A Japanese study performed whole exome sequencing of the ErbB4 protein and documented ALS symptoms in patients with a genetic mutation (p. Arg927Gln) within the tyrosine kinase domain, which mediates the function of the protein [12]. Likewise, three possibly pathologic variants were also documented in a Chinese cohort (p. Arg106His, p. Gln164Pro, and p. Val212Leu) that did not carry common pathogenic mutations related to ALS such as SOD1, TARDBP, FUS, and C9ORF72. These variants are located upstream of the tyrosine kinase encoding region of the gene [12-13]. In its supplementary data, the same chromosomal DNA mutation (p. Arg979Gln) was documented in 3 of its 1812 controls who did not manifest with any neurological symptoms at the time of onset of the study, and none in the 448 patients who had neurologic symptoms [13]. This offers, then, the possibility that this variant, though still documented as of uncertain significance, can offer insight into the pathogenicity of ALS in patients with movement disorders, and is worth studying. Its implication may also contribute to the rapidity of disease development or even a predominant motor neuron disease presentation in patients with Parkinsonism.
Conclusions
Despite the great lengths clinical and genetic research has gone, a diagnostic dilemma still exists in patients with movement disorders. Diagnostic modalities can still bolster the primary impression despite both clinical acumen and exhaustive physical examination. In this patient, an index of suspicion guided clinicians to a diagnosis, but required other modalities to further confirm that both central and peripheral neurodegenerative diseases coexisted in this patient. This emphasizes the armamentarium of means a neurologist can use in patient care, from one's own senses to ancillary diagnostics. Furthermore, this case shows the difficulties that can be encountered in treating neurodegenerative movement disorders.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Addressing the role of edaravone in the management of amyotrophic lateral sclerosis and gaps in care and access: expert panel recommendations Am J Manag Care Barrington CJ Burruano M Carney C 07272021 https://doi.org/10.37765/ajmc.2021.8873210.37765/ajmc.2021.8873234382759 · doi ↗ · pubmed ↗
- 2Effects of the edaravone, a drug approved for the treatment of amyotrophic lateral sclerosis, on mitochondrial function and neuroprotection Antioxidants (Basel) Cha SJ Kim K 195112022 http://dx.doi.org/10.3390/antiox 110201953520407810.3390/antiox 11020195 PMC 8868074 · doi ↗ · pubmed ↗
- 3Multiple system atrophy Int Rev Neurobiol Fanciulli A Stankovic I Krismer F Seppi K Levin J Wenning GK 1371921492019 https://doi.org/10.1016/bs.irn.2019.10.0043177981110.1016/bs.irn.2019.10.004 · doi ↗ · pubmed ↗
- 4Atypical parkinsonism of Japan: amyotrophic lateral sclerosis-parkinsonism-dementia complex of the Kii peninsula of Japan (Muro disease): an update Mov Disord Kuzuhara S Kokubo Y 013202005 https://doi.org/10.1002/mds.2054810.1002/mds.2054816092099 · doi ↗ · pubmed ↗
- 5Improving diagnostic accuracy of multiple system atrophy: a clinicopathological study Brain Miki Y Foti SC Asi YT Tsushima E Quinn N Ling H Holton JL 281328271422019 https://doi.org/10.1093/brain/awz 1893128981510.1093/brain/awz 189 · doi ↗ · pubmed ↗
- 6Coenzyme Q 10 in multiple system atrophy Trials for Cerebellar Ataxias. Contemporary Clinical Neuroscience Mitsui J Tsuji S 679690 Cham, Switzerland Springer 2023 https://doi.org/10.1007/978-3-031-24345-5_28
- 7Investigating edaravone use for management of amyotrophic lateral sclerosis (ALS): a narrative review Cureus Neupane P Thada PK Singh P 0152023 https://doi.org/10.7759/cureus.3374610.7759/cureus.33746 PMC 992252336788871 · doi ↗ · pubmed ↗
- 8Neuregulins in neurodegenerative diseases Front Aging Neurosci Ou GY Lin WW Zhao WJ 662474132021 https://doi.org/10.3389/fnagi.2021.6624743389740910.3389/fnagi.2021.662474 PMC 8064692 · doi ↗ · pubmed ↗