Acute administration of lovastatin had no pronounced effect on motor abilities, motor coordination, gait nor simple cognition in a mouse model of Angelman syndrome
Timothy A. Fenton, Stela P. Petkova, Anna Adhikari, Jill L. Silverman

TL;DR
A study found that lovastatin, a drug with low toxicity and cost, did not improve symptoms in a mouse model of Angelman syndrome and even worsened outcomes in control mice.
Contribution
The study evaluates the effects of acute lovastatin administration in Angelman syndrome mouse models, revealing unexpected negative behavioral outcomes.
Findings
Acute lovastatin administration worsened exploratory activity in Angelman syndrome mice and sedated control mice.
Lovastatin did not improve motor coordination, gait, or cognition in Angelman syndrome mice.
Cognitive performance in a novel object recognition task was worsened in wildtype controls after lovastatin treatment.
Abstract
Translational research is needed to discover pharmacological targets and treatments for the diagnostic behavioral domains of neurodevelopmental disorders (NDDs), including autism spectrum disorders (ASDs) and intellectual disabilities (IDs). One NDD, associated with ASD and ID, is Angelman Syndrome (AS). AS is a rare genetic NDD for which there is currently no cure nor effective therapeutics. The genetic cause is known to be the loss of expression from the maternal allele of ubiquitin protein ligase E3A (UBE3A). The Ube3a maternal deletion mouse model of AS reliably demonstrates behavioral phenotypes of relevance to AS and therefore offers a suitable in vivo system in which to test potential therapeutics, with construct and face validity. Successes in reducing hyperexcitability and epileptogenesis have been reported in an AS model following acute treatment with lovastatin, an ERK…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
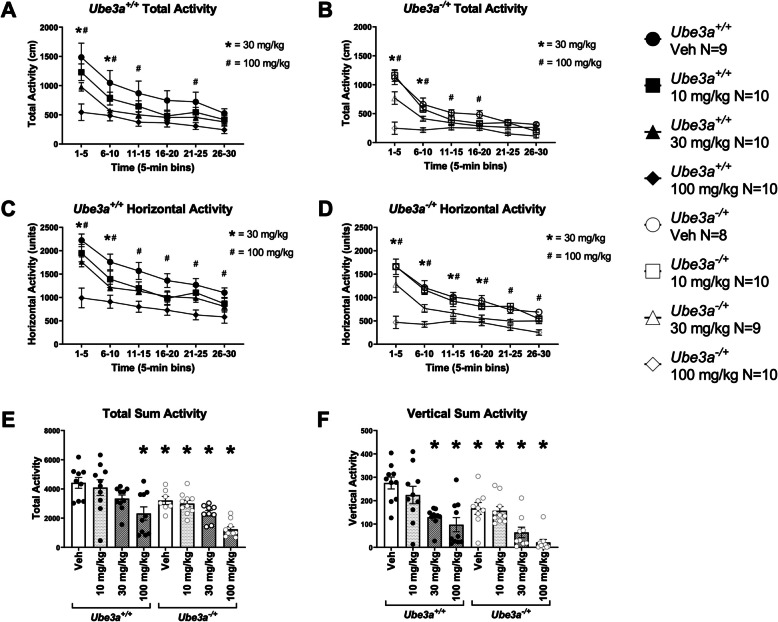 Figure 1
Figure 1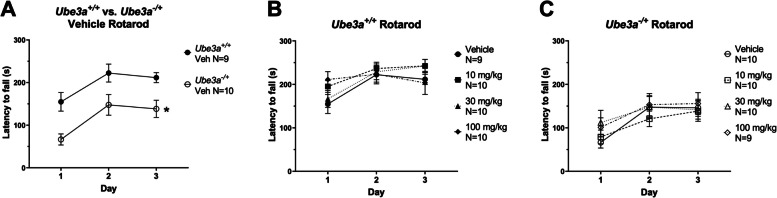 Figure 2
Figure 2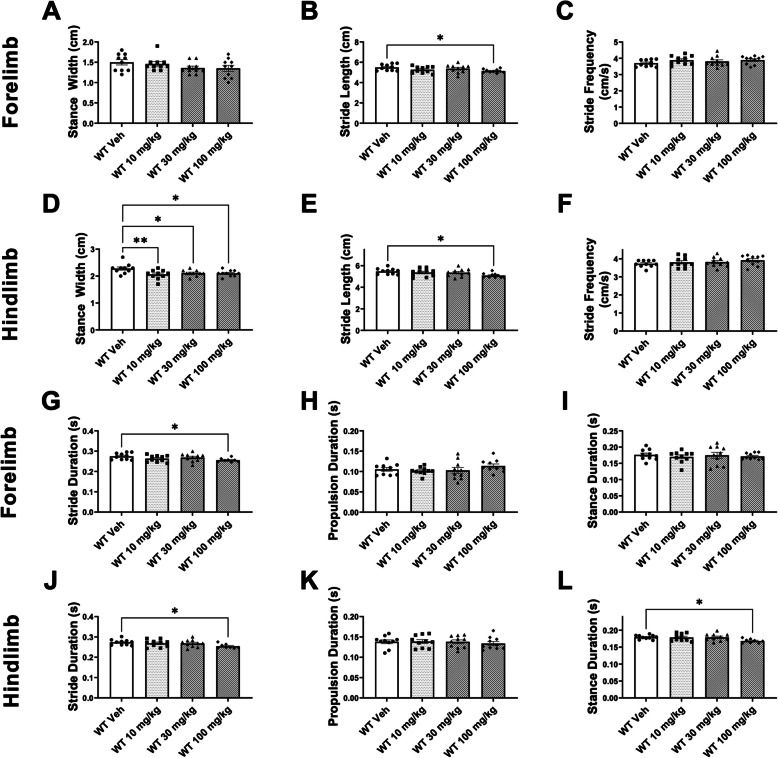 Figure 3
Figure 3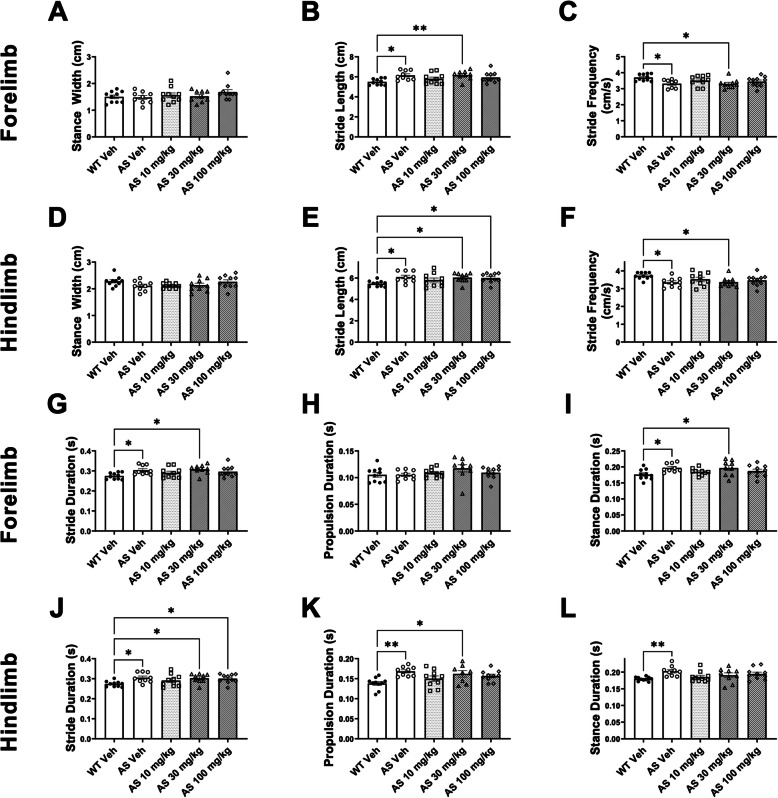 Figure 4
Figure 4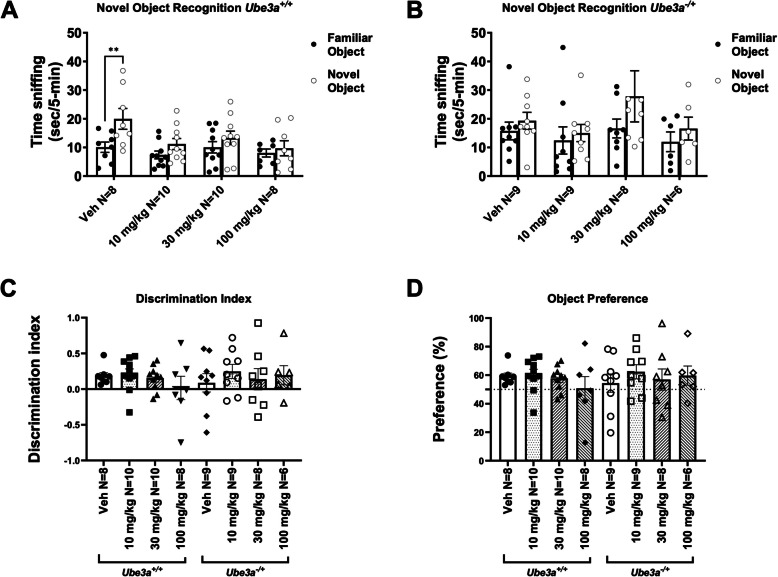 Figure 5
Figure 5- —http://dx.doi.org/10.13039/100009633Eunice Kennedy Shriver National Institute of Child Health and Human Development
- —http://dx.doi.org/10.13039/100009726Foundation for Angelman Syndrome Therapeutics
- —NIH/NINDS
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsGenetic Syndromes and Imprinting · Growth Hormone and Insulin-like Growth Factors · Cancer-related molecular mechanisms research
Introduction
Angelman Syndrome (AS) is a rare neurodevelopmental disorder (NDD) caused by the loss of functional ubiquitin protein ligase E3A (UBE3A) [1–3]. Specifically, AS results from a loss of expression from the maternal allele, leaving the brain deficient of UBE3A in most brain regions, because of neuronal-specific imprinting that silences the paternal allele with the minor exception of the superchiasmatic nucleus (SCN) [4–6] and contributions from glia, illustrated previously in AS mouse models, primates and humans [7, 8]. AS is characterized by developmental delay, intellectual disability (ID), impaired communication, gross and fine motor deficits, including movement, coordination, balance and gait, seizures and abnormal sleep [9–12]. Since these symptoms are severe and persistent, and there is currently no effective therapeutic or cure for the disorder, those with AS require lifelong supportive care. It is therefore imperative that novel strategies to treat AS are developed.
While precision therapy approaches offer future potential for disease-modifying treatments of genetically defined disorders, our understanding of these valuable therapies is that UBE3A is critical for postnatal brain development and plays a critical role in multiple neuronal signaling pathways [13]. Unfortunately, our understanding of these valuable therapies is relatively new. At the time of writing, only three gene editing therapies for neurological disorders were currently FDA approved [14]. Several molecular approaches to restore UBE3A expression in the brain are being developed including: microRNA with adeno associated viral vectors [15, 16], CRISPR/Cas9 gene therapy [17], replacement via a lentiviral replacement of a healthy Ube3a copy via hemopoietic stem cells [18], artificial transcription factors (ATFs) [19–22], and antisense oligonucleotides (ASOs) [23–25], to mediate paternal activation. Additional ASOs are currently being modified by industry to generate novel molecules that are being evaluated in clinical trials NCT04259281/Ultragenyx). These translational approaches have been supported by numerous basic science studies yet more research focused on the consequences of UBE3A restoration in the brain is required.
Pharmacological therapies, while not disease modifying, can have a great impact on specific symptom domains, such as motor abilities, sleep, and/or seizures, significantly improving the quality of life of individuals with AS, their families and caregivers. There is an ever-growing list of repurposed compounds that have been suggested to improve phenotypes in AS mouse models. Among these are IGF2 [26, 27], the histone deacetylation inhibitor sodium valproate [28], channel inhibitors [29], and protein phosphatase 2A inhibitor LB-100 [30]. In many rescue reports, the primary assays have been the electrophysiological rescue of long-term potentiation (LTP) in hippocampal slices. The persistent strengthening of synapses via LTP is dogmatically considered the underlying mechanism of action of learning and memory, yet many compounds that rescue LTP have not proven to be efficacious cognitive enhancers in vivo [29, 31–33].
Here we performed a tailored set of behavioral assays translationally relevant to AS, to examine doses of lovastatin in vivo. We focused on assays of motor and cognition, as these have been well reproduced clinically and are profoundly impaired clinically.
Recently, there has been wide interest in the use of statins to treat NDDs [34, 35]. Specific to AS, neurons from AS models have excessive levels of synaptic proteins, thus reducing protein synthesis with lovastatin may have clinical benefit [13, 36]. Additional rationale that guided our study of lovastatin includes its earlier success in AS mice reversing LTP impairments and improving audiogenic seizures [37]. Lovastatin also showed improvements in a variety of domains in the FXS mouse model such as reduction in both protein synthesis and audiogenic seizures [38–41] and improvements in cognition in the FXS rat model [42]. As many of these domains of behavioral deficiency in FXS overlap with AS, our laboratory assessed several doses of lovastatin as a potential therapeutic for AS in vivo using a behavioral battery relevant to AS that have been previously published [18, 22, 43–47].
Repurposing lovastatin for the treatment of AS, is an attractive prospect since it is currently widely prescribed, has been shown to be safe and well tolerated, and is approved for use in children [48]. These factors could drastically reduce the financial burden and time it takes for a drug to be used in the clinic for AS. Even if only a small subset of symptoms is alleviated, this safe, repurposed therapeutic could reduce immense hardship that individuals with AS, caregivers and their families face while waiting for a novel, curative, gene editing therapy. Thus, our objectives in this report were 1) to pharmacologically assess a variety of doses of lovastatin as a potential therapeutic for AS using an in vivo model with robust behavioral phenotypes and their WT age and sex matched controls, 2) confirm the most effective acute dose in AS models that was not deleterious and led to functional improvements, and 3) identify any adverse or deleterious effects of lovastatin administration in WT and sex and age matched AS experimental models.
Materials and methods
Mice for behavioral testing
All animals were housed in Techniplast cages (Techniplast, West Chester, PA, USA) in a temperature (68–72 °F) and humidity (~ 25%) controlled vivarium maintained on a 12:12 light–dark cycle. All procedures were approved by the Institutional Animal Care and Use Committee (IACUC) at the University of California Davis and were conducted in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. B6.129S7-Ube3a^tm1 Alb/J^ mice were obtained from The Jackson Laboratory (Bar Harbor, ME, USA) and fed a standard diet of Teklad global 18% protein rodent diets 2918 (Envigo, Hayward, CA, USA). Rodent chow and tap water were available ad libitum. In addition to standard bedding, a Nestlet square, shredded brown paper, and a cardboard tube (Jonesville Corporation, Jonesville, MI) were provided in each cage. Heterozygous deletion female mice were bred with C57BL/6 J (B6 J) male mice to generate maternal deletion (Ube3a^−/+^, AS) and wildtype (Ube3a^+/+^, WT) littermates. To identify mice, pups were labeled by paw tattoo over postnatal days (PND) 2–4 using non-toxic animal tattoo ink (Ketchum Manufacturing Inc., Brockville, ON, Canada). Tails of pups were clipped (1–2 mm) for genotyping, following the UC Davis IACUC policy regarding tissue collection. Genotyping was performed with REDExtract-N-Amp (Sigma Aldrich, St. Louis, MO, USA) using primers JAX 25383 TCAATGATAGGGAGATAAAACA, 25384 GAAAACACTAACATGGAGCTC, and 25385 CTTGTGTAGCGCCAAGTGC.
To reduce carry-over effects from repeated behavioral testing, at least 48 h were allowed to pass between the completion of one task, and the start of the next task in the order of the behavioral battery [18, 26, 47, 49]. The interval between behavioral testing also allowed for the clearance of any residual lovastatin which has a half-life of 0.7–3 h [50, 51]. On days between behavioral tasks, animals were not injected. Assays were performed in the order of least to most stressful, as previously published, and between the hours of 8:00 AM PST and 7:00PM PST, during the light phase. Group sizes were chosen based on experience and power analyses [52–60].
All behavior testing was conducted by an experimenter that was blind to both genotype and treatment and included both sexes. Examining sex as a biological variable is typically, the standard practice in our laboratory. In the case of AS rodent models, our laboratory has performed analysis of sex differences over several years, across a variety of studies in AS rat and mouse models. We discovered no significant sex differences in AS rodents in the open field, the rotarod, spatial or temporal metrics of gait, the novel object recognition task, EEG, nor location discrimination and pairwise discrimination, touchscreen assays of cognition [26, 43–47, 53, 61, 62]. Other laboratories have also failed to observe sex differences in AS mice and rats, compared to WT [45, 63–66]. Cohorts were comprised of both sexes were utilized to identify any unpredicted sex effects, resulting from lovastatin treatment.
Mice were allowed to habituate in their home cages in a dimly lit room adjacent to the testing room for 1 h prior to the start of testing to limit any effect of transporting between the vivarium and testing suite. Between mice, testing apparatus surfaces were cleaned using 70% ethanol and allowed to dry. One cohort of animals was tested comprised of 14 litters beginning at 8 weeks of age (PND 55). To avoid any effect of litter, animals from each treatment group originated from between 5 and 8 different litters with no more than 3 mice from the same litter being included in any single treatment group. The order of testing was (1) open field, (2) accelerating rotarod, (3) DigiGait, (4) spontaneous alternation in the y-maze, and (5) novel object recognition. Weights of each mouse were taken prior to injection on each day that a behavior task was executed.
Drug preparation
Lovastatin (TCI America™, L0214) was dissolved in Caprylic/Capric Triglyceride (Spectrum Chemical, C3465) at the beginning of each behavioral testing day. At the beginning of the study animals of each genotype were randomly assigned to different groups to determine which dose they would receive throughout the length of the study. Each mouse was injected IP with 10 mg/kg, 30 mg/kg, or 100 mg/kg of lovastatin or vehicle prior to performing each behavioral task. Doses were chosen based previous research in NDDs, such as Li and Silva in Neurofibromitosis-1 (NF1), Osterweil and Bear, and Muscas and Osterweil in Fragile X Syndrome (FXS), and Chung and Jiang’s report in Angelman Syndrome [37, 39, 40, 67]. To control for the half-life of lovastatin, injections were performed with staggered timing to ensure each subject was injected exactly 1 h before starting the behavioral task. In the case of multi-day tasks, subjects were injected on each day of the task, 1 h prior to beginning the task [68–71].
Open field
General exploratory locomotion in a novel open field environment was assayed in an arena sized 40 cm × 40 cm × 30.5 cm, as previously described [22, 47, 49, 72–77]. Open field activity was considered an essential control for effects on physical activity, for example, sedation or hyperactivity which could confound the interpretation of result of interaction with objects, arena, and object exploration, and sniffing and investigation times in subsequent behavioral assays. The testing room was illuminated at ~ 40 lx.
Rotarod
Motor coordination, balance, and motor learning were assessed using an accelerating rotarod (Ugo Basile, Schwenksville, PA) as previously described [18, 26, 47]. The task requires the mice to walk forward to remain on top of the rotating cylinder rod. Mice were given 3 trials per day with a 30–60-min inter-trial rest interval. Mice were tested over 3 consecutive days for a total of 9 trials. Latency to fall was recorded with a 300-s maximum latency.
Gait
Treadmill gait analysis was performed using the DigiGait™ system (Mouse Specifics Inc., USA), as described [47]. Before data collection, each subject was acclimated to the Perspex walking chamber for 1 min and the treadmill was slowly accelerated to the final speed of 20 cm/s to allow mice to adjust to walking on the belt. Digital images of paw placement were recorded through a clear treadmill from the ventral plane of the animal. Mice were tested in a single session at a 20 cm/s treadmill speed maintaining a normal pace walk for Ube3a^+/+^ mice. Non-performers were defined as mice who were unable to sustain walking at 20 cm/s without colliding with the posterior bumper for at least 3 s. There is no practice effect, therefore, mice were allowed retrial and retest if they were unable to adjust to walking on the belt easily. The treadmill belt and the encasing Perspex chamber were cleaned with 70% (v/v) ethanol in between mice. For each mouse, videos of ∼5 s duration of all sessions were analyzed using the DigiGait™ Imaging and Analysis software v12.2 (Mouse Specifics Inc., USA). Contrast filters were determined on a mouse-by-mouse case to facilitate consistent recognition of all four paws. All analysis was conducted in a single session by an experimenter blind to genotype. Stance width (distance between paws), stride duration (time between heel strikes of the same paw), propulsion duration (time from full stance to toe push off), stance duration (time paw is in contact with the ground), stride length (distance a paw makes during a single stride) and stride frequency (number of strides per second to maintain pace) were automatically calculated. Data was averaged between left and right paws.
Spontaneous alternation
Spontaneous alternation in a Y-maze was assayed using methods modified from previous studies [18, 26, 60, 73, 78, 79]. The Y-maze assesses spontaneous alternation and can assess short-term spatial memory [80–82]. Performance is dependent on an intact hippocampus and various interconnected structures [83]. When placed within a maze with multiple arms, mice exhibit the spontaneous behavior of alternating between arm choices with a greater frequency than re-entering the same arm most recently visited [80, 84, 85]. Our apparatus and protocol were internally validated using anticholinergics, such as scopolamine, which have shown attenuation in Y-maze alternation performance, and thus we used scopolamine as a negative control, and a task validation tool. As scopolamine is a non-specific drug, we limited inferences on “working memory.” In the absence of a proven cognitive enhancer to provide a positive control for our work, and the lack of consensus on what/when “working memory” ends, we have steered away from any controversial terminology or overinterpretation of the Y-maze assay, using “spontaneous alternation memory in the Y-maze.” Mice explored a Y- shaped maze constructed of matte white acrylic (P95 White, Tap Plastics, Sacramento, CA, USA) for 8 min and were recorded from an overhead camera with the behavioral tracking software Ethovision XT. Mice were placed at the center of the initial arm facing the center of the maze. Percentage of spontaneous alternations is calculated as the number of triads (entry into three different arms without returning to a previously entered arm) relative to the number of alternation opportunities. All scoring was conducted by an observer blind to genotype and treatment.
Novel object recognition
The novel object recognition (NOR) test was conducted in opaque matte white (P95 White, Tap Plastics, Sacramento, CA) open field arenas (41 cm × 41 cm × 30 cm), using methods similar to those previously described [18, 49, 73–75, 86]. The experiment consisted of 4 sessions: a 30-min exposure to the open field arena the day before the test, a 10-min re-habituation on test day, a 10-min familiarization session and a 5-min recognition test. On day 1, each subject was habituated to a clean, empty, open field arena for 30-min. 24-h later, each subject was returned to the open field arena for another 10- min for the habituation phase. The mouse was then removed from the open field and placed in a clean temporary holding cage for approximately 2-min. Two identical objects were placed in the arena. Each subject was returned to the open field in which it had been habituated and allowed to freely explore for 10-min. After the familiarization session, mice were returned to their holding cages, which were transferred from the testing room to a nearby holding area for an interval of 60 min. The open field was cleaned with 70% ethanol and allowed to dry. One clean familiar object and one clean novel object were placed in the arena, where the two identical objects had been located during the familiarization phase. Each subject was returned to its open field for a 5-min recognition test, during which time it was allowed to freely explore the familiar object and the novel object. The familiarization session and the recognition test were recorded with Ethovision XT video tracking software (Version 9.0, Noldus Information Technologies, Leesburg, VA) and manually scored by an observer blinded to genotype and drug treatment. Object investigation was defined as time spent sniffing the object when the nose was oriented toward the object and the nose–object distance was 2-cm or less. Recognition memory was defined as spending significantly more time sniffing the novel object compared to the familiar object determined within genotype and within dose. Total time spent sniffing both objects was used as a measure of general exploration. Time spent sniffing two identical objects during the familiarization phase confirmed the lack of an innate side bias. Objects used were plastic toys: a small soft plastic orange safety cone and a hard plastic magnetic cone with ribbed sides, as previously described [86].
Statistical analysis
Data were analyzed with GraphPad Prism 9 (GraphPad Software, San Diego, CA). Sex differences have not been observed in any assay, using this tailored behavioral battery for AS to date, and thus we combined sexes were utilized to achieve a powerful sample size [18, 26, 47]. Behavioral analysis passed normality distribution, using D’Agostino and Pearson tests. Data was collected using continuous variables, and thus, was analyzed via parametric analysis. Outliers were identified and excluded using Grubb’s test. Statistical testing was performed using established assay-specific methods, including two tailed student’s t-test for single parameter comparisons for spontaneous alternation and novel object recognition tasks. Where appropriate, one-way, or two-way ANOVA tests were used for open field, rotarod, and DigiGait™. As time was a variable for the open field and rotarod, two-way repeated measures ANOVA were used. Significant ANOVAs were followed by multiple comparisons, using Dunnett’s post hoc testing, when lovastatin treated groups were compared to vehicle treated groups. All significance levels were set at p < 0.05. Data are presented as mean ± standard error of the mean (S.E.M.) unless otherwise noted.
Results
Lovastatin reduced exploration in Ube3a+/+ (WT) and Ube3a−/+ (AS) mice
Motor function is highly translational and consists of many nuanced components, including gross exploratory locomotion, movement distances, and abilities, such as balance, coordination, and gait. We utilized a tailored motor battery to perform a motor assessment that may be useful for clinical trials, which included novel locomotion in an open field arena, the accelerating rotarod, and gait analysis. As expected, we observed vehicle treated mice of both genotypes, Ube3a^+/+^ wildtype (WT) and Ube3a^−/+^ (AS), habituate to the open field arena via reduced total activity, when compared by two-way repeated measures ANOVA for genotype and lovastatin, at different doses over time (F time x genotype x dose (35, 345) = 4.736, p < 0.0001). Lovastatin had effects in WT and AS mice (Fig. 1A; F dose inUbe3a^+/+^(3, 36) = 4.156, p < 0.02; Fig. 1B; F dose inUbe3a^−/+^(3, 35) = 4.949, p < 0.0005), in the total distance metric. We also observed vehicle treated mice of both genotypes habituate to the open field arena, via reduced horizontal activity, when compared by two-way repeated measures ANOVA for genotype and lovastatin, at different doses, over time (F time x genotype x dose (35, 345) = 3.913, p < 0.0001). Genotype effects showed that AS mice differed from WT (F genotype x dose (7, 69) = 13.70, p < 0.0001). Lovastatin had effects in WT and AS mice (Fig. 1C; F dose inUbe3a^+/+^(3, 36) = 7.238, p < 0.0001; Fig. 1D; F dose inUbe3a^−/+^(3, 36) = 11.41, p < 0.0005), on the horizontal activity metric. Supplementary Tables S1 and S2 illustrate the post hoc analysis. AS mice treated with vehicle traveled less distance in the open field, compared to vehicle treated WT mice (Fig. 1E; F genotype (1, 68) = 20.30, p < 0.0001), when all time-bins were summed.Fig. 1. Lovastatin reduced exploration in Ube3a^+/+^ and Ube3a^−/+^ mice. A, C Lovastatin induced dose dependent decreases in locomotor activity in Ube3a^+/+^ mice in both (A) total activity and (C) horizontal activity, over time. Lovastatin, 30 mg/kg and 100 mg/kg reduced (B) total activity and (D) horizontal activity, over time in Ube3a^−/+^ mice. E Lovastatin at the 100 mg/kg dose caused a significant reduction in summed total activity across a 30-min session in both Ube3a^+/+^ and Ube3a^−/+^ mice. Lovastatin, 10 mg/kg and 30 mg/kg also reduced summed total activity in Ube3a^−/+^ mice. F Lovastatin, 10 mg/kg and 30 mg/kg reduced summed vertical activity (rearing) in Ube3a^+/+^ mice. Ube3a^−/+^ mice also displayed reduced summed vertical activity when treated with vehicle or any dose of lovastatin, compared to vehicle treated Ube3a^+/+^. * p < 0.05, A-D, two-way repeated measures ANOVA over time by 30 mg/kg of lovastatin and genotype. # p < 0.05, A-D, two-way repeated measures ANOVA over time by 100 mg/kg of lovastatin and genotype. * p < 0.05, E and F, one –way ANOVA for lovastatin, followed by Holm-Sidak post hoc analysis
Lovastatin treatment reduced summed total activity in WT treated with 100 mg/kg of lovastatin (Fig. 1E; F dose inUbe3a^+/+^ (3, 36) = 1.428, p < 0.0001). Lovastatin also reduced summed vertical activity in WT mice (Fig. 1F; F dose inUbe3a^+/+^(3, 36) = 1.981, p < 0.0001), treated with 30 mg/kg (p < 0.0005) and 100 mg/kg (p < 0.0001), using a one-way ANOVA, and Holm-Sidak’s post hoc analysis for multiple comparisons. Over time, and unexpectedly, detrimental effects of lovastatin were observed in WT subject mice during the total activity assessment (Fig. 1A; F dose inUbe3a^+/+^(3, 36) = 4.156, p < 0.02). Dunnet’s post hoc analysis illustrated an adjusted p value of p = 0.001, comparing 100 mg/kg versus WT veh (Bin 1–5 min), an adjusted p value of p = 0.013, comparing 30 mg/kg versus WT veh, and p = 0.0027, comparing 100 mg/kg versus WT veh (Bin 6–10 min). Dunnet’s post hoc analysis illustrated an adjusted p value of p = 0.0095, comparing 100 mg/kg versus WT veh (Bin 11–15 min), and an adjusted p value of p = 0.0369, comparing 100 mg/kg versus WT veh (Bin 21–25). These data were calculated using a one-way ANOVA, and Holm-Sidak’s post hoc analysis for multiple comparisons (Supplementary Table S4).
Additionally, over time, and unexpectedly, detrimental effects of lovastatin were observed in WT subject mice when using the horizontal activity parameter (Fig. 1C; F dose inUbe3a^+/+^(3, 36) = 7.238, p < 0.0001). Dunnet’s post hoc analysis illustrated an adjusted p value of p = 0.001, comparing 30 mg/kg versus WT veh, and p < 0.0001, comparing 100 mg/kg versus WT veh (Bin 1–5 min), an adjusted p value of p = 0.0065, comparing 100 mg/kg versus WT veh (Bin 6–10 min). Dunnet’s post hoc analysis illustrated an adjusted p value of p = 0.0152, comparing 100 mg/kg versus WT veh (Bin 11–15 min), an adjusted p value of p = 0.0161, comparing 100 mg/kg versus WT veh (Bin 16–20), an adjusted p value of p = 0.0082, comparing 100 mg/kg versus WT veh (Bin 21–25), and an adjusted p value of p = 0.0335, comparing 100 mg/kg versus WT veh (Bin 26–30). These data were calculated using a one-way ANOVA, and Holm-Sidak’s post hoc analysis for multiple comparisons (Supplementary Table S6).
AS mice, treated with each dose of lovastatin, (Fig. 1E; F dose inUbe3a^−/+^(3, 36) = 1.185, 10 mg/kg p < 0.1232, 30 mg/kg p < 0.00097 and 100 mg/kg p < 0.0001), exhibited reduced summed total activity compared to vehicle treated WT mice using a one-way ANOVA, and Holm-Sidak’s post hoc analysis for multiple comparisons (Supplementary Tables S5). Genotype reduced summed vertical activity (i.e., rearing) in vehicle treated AS mice, compared to vehicle treated WT mice, (Fig. 1F; F genotype (1, 68) = 19.36, p < 0.0001). In AS mice, every dose of lovastatin, reduced summed vertical activity (Fig. 1F; F dose inUbe3a^+/+^(3, 34) = 12.96, 10 mg/kg p < 0.1232, 30 mg/kg p < 0.00097 and 100 mg/kg p < 0.0001), using a one-way ANOVA, and Holm-Sidak’s post hoc analysis for multiple comparisons (Supplementary Table S7).
Lovastatin did not adversely affect rotarod performance in WT mice, but did not improve the impaired rotarod performance of AS mice
A corroborating assay of motor abilities, coordination, and motor learning is the accelerating rotarod. We observed intact performance over time in Ube3a^+/+^ and Ube3a^−/+^ mice*,* over the three training days (Fig. 2A; F time (2, 50) = 8.861, p < 0.0005), a genotype effect was present in the vehicle treated Ube3a^−/+^ group with significantly reduced latencies to fall compared to vehicle treated mice of the Ube3a^+/+^ group (Fig. 2A; F genotype (1, 50) = 25.02, p < 0.0001). Training day effects were observed on the rotarod in Ube3a^+/+^ mice (Fig. 2B; F time inUbe3a^+/+^ (2, 105) = 7.967, p < 0.0006). No significant main effect of lovastatin was observed on the rotarod in Ube3a^+/+^ mice using a dose by repeated measures ANOVA statistic (Fig. 2B; F dose x time inUbe3a^+/+^ (3, 105) = 1.202, NS). Ube3a^−/+^ mutant mice displayed motor learning, via a significant main effect of training day, using a repeated measures ANOVA (Fig. 2C; F time inUbe3a^−/+^ (2, 105) = 7.992, p < 0.0006). No significant effects of lovastatin were observed on the rotarod in Ube3a^−/+^ mice using a dose by repeated measures ANOVA (Fig. 2C; F dose x time inUbe3a^−/+^ (3, 105) = 0.7801, NS).Fig. 2. Lovastatin did not affect rotarod performance in Ube3a^+/+^ and Ube3a^−/+^ mice. A No significant effect of lovastatin was observed on the rotarod in Ube3a^+/+^ mice. B No significant effect of lovastatin was observed on the rotarod in Ube3a-/+ mice. C Vehicle treated Ube3a^−/+^ mice displayed significantly reduced latencies to fall when compared to Ube3a^+/+^ mice, at each time point, * p < 0.05, repeated-measures two-way ANOVA, followed by Holm-Sidak post-hoc analysis
Lovastatin disrupted spatial and temporal gait parameters in WT mice
We observed unexpected alterations of several spatial gait metrics, assayed using the DigiGait treadmill. Lovastatin treatment (100 mg/kg) reduced forelimb and hindlimb stride length in WT subject mice compared to vehicle treatment (Figs. 3B, E). Hindlimb stance width was reduced in WT mice, when treated with 10 mg/kg, 30 mg/kg or 100 mg/kg of lovastatin (Fig. 3D), when analyzed by a one-way ANOVA, at each dose evaluated (Fig. 3D; F dose inUbe3a^+/+^(3, 36) = 2.621, p < 0.05), followed by Dunnett’s post hoc analysis (10 mg/kg, p < 0.030; 30 mg/kg, p < 0.040; 100 mg/kg, p < 0.030). In WT mice, forelimb and hindlimb stride duration were reduced by 100 mg/kg of lovastatin (Figs. 3G, J). 100 mg/kg of lovastatin, reduced forelimb (Fig. 3G; F dose inUbe3a^+/+^(3, 36) = 2.377, p < 0.05) and hindlimb stride duration (Fig. 3J; F dose inUbe3a^+/+^(3, 36) = 1.857), analyzed by a one-way ANOVA, followed by Dunnett’s post hoc analysis (100 mg/kg forelimb, p < 0.0429 and 100 mg/kg hindlimb, p < 0.0345). Hindlimb stance duration in WT mice*,* treated with 100 mg/kg of lovastatin, was also reduced (Fig. 3L; F dose inUbe3a^+/+^(3, 36) = 3.614, p < 0.03), when compared using a one-way ANOVA, followed by Dunnett’s post hoc analysis (100 mg/kg hindlimb stance duration, p < 0.0455). Multiple parameters indicated no significant differences resulting from lovastatin treatment in forelimb and hindlimb stride frequency (Figs. 3 C, F), forelimb stance width and propulsion duration (Figs. 3A, I), and hindlimb propulsion duration (Fig. 3K).Fig. 3. Lovastatin disrupted spatial and temporal gait parameters in Ube3a^+/+^ mice. Sedating effects of lovastatin on metrics of gait in Ube3a^+/+^ mice (WT). Lovastatin treatment at any dose did not affect (A) forelimb stance width, but all doses of lovastatin reduced the (D) hindlimb stance width. Both (B) forelimb stride length and (E) hindlimb stride length were significantly reduced when Ube3a^+/+^ mice were treated with 100 mg/kg of lovastatin. No changes were observed in (C) forelimb stride frequency or (F) hindlimb stride frequency at any dose. Reductions in (G) forelimb stride duration and (J) hindlimb stride duration were present when treated with 100 mg/kg of lovastatin. No changes were observed at any dose of lovastatin in the (H) forelimb propulsion duration or (K) hindlimb propulsion duration. Finally, a reduction in (L) hindlimb stance duration was observed, following 100 mg/kg of lovastatin, but no changes were observed in the (I) forelimb stance duration. Number of mice tested for gait: WT Veh N = 10, 10 mg/kg N = 8, 30 mg/kg N = 8, 100 mg/kg N = 7; AS Veh N = 9, 10 mg/kg N = 10, 30 mg/kg N = 9, 100 mg/kg N = 10. * p < 0.05, two-way ANOVA, one–way ANOVA followed by Dunnett’s post hoc analysis
Lovastatin did not improve spatial and temporal gait parameters in AS mice
We observed no effects of lovastatin treatment in forelimb nor hindlimb stance width (Figs. 4A, D), nor forelimb propulsion duration (Fig. 4H) in AS mice. For forelimb and hindlimb stride length, genotype and lovastatin dose effects were identified using two-way ANOVAs (forelimb: Fig. 4B; F genotype (1, 67) = 56.12, p < 0.001; Fig. 4B; F dose inUbe3a^−/+^(3, 37) = 3.419, p = 0.0163 and hindlimb: Fig. 4E; F genotype (1, 67) = 59.78, p < 0.0001; Fig. 4E; F dose inUbe3a^−/+^(3, 37) = 2.912, p < 0.05). Vehicle treated AS mice differed from vehicle treated WT mice in forelimb and hindlimb stride length, stride frequency and stride duration (Figs. 3B, C, E, F, G, J), forelimb stance duration (Fig. 3I), and hindlimb propulsion and stance duration (Figs. 3K, L). AS mice had extended stride length, stride and stance duration, and hindlimb propulsion duration, and exhibited less stride frequency compared to WT mice. In some metrics such as stride length, lovastatin treated AS mice did not differ from vehicle treated WT mice, but also did not differ from AS vehicle mice, and thus we attributed these effects to lovastatin treatment as potentially mild, but inconclusive gait improvements. It is worth noting that differences in stride length between lovastatin treated AS mice and AS vehicle treated mice, were not detectable. Doses of 10 mg/kg (p = 0.5825) and 100 mg/kg (p = 0.2072) of lovastatin treatment in AS mice exhibited reduced forelimb stride lengths, making these AS groups indistinguishable from vehicle treated WT. Yet, despite not being significantly different from vehicle treated WT mice, differences between lovastatin treated AS mice and the forelimb stride length of the AS vehicle treated mice were not detectable, which suggests a subtle effect. 30 mg/kg, and 100 mg/kg of lovastatin treatment in AS mice resulted in extended hindlimb stride length, indistinguishable from vehicle treated AS mice, but increased compared to vehicle treated WT mice, (30 mg/kg, p < 0.0367 and 100 mg/kg, p < 0.0475). Genotype and lovastatin dose effects, respectively, were observed in forelimb and hindlimb stride frequency, using two-way ANOVAs (forelimb: Fig. 4C; F genotype(1, 67) = 53.75, p < 0.001 and F dosein Ube3a^−/+^(3, 67) = 2.855, p < 0.05 and hindlimb: Fig. 4F; F genotype (1, 67) = 48.73, p < 0.001 and F dose inUbe3a^−/+^(3, 67) = 2.269, p < 0.05). Stride frequency is reduced in AS mice compared to WT mice, by the forelimb and hindlimb indices when treated with vehicle (Fig. 4C, F). AS mice treated with 30 mg/kg of lovastatin exhibited reduced forelimb and hindlimb stride frequency compared to vehicle treated WT mice, but not versus vehicle treated AS mice following Dunnett’s post hoc analysis (30 mg/kg forelimb: p < 0.0140 and 30 mg/kg hindlimb: p < 0.0351). Although AS mice treated with 10 mg/kg or 100 mg/kg exhibit stride frequencies that are not significantly lower from vehicle treated WT mice, this fails to illustrate a robust effect of lovastatin treatment because of the lack of difference to vehicle treated AS mice. Stride length and frequency are the two metrics that have previously been reported to be altered in AS rodent models and individuals with AS by independent groups [26, 47, 87–89].Fig. 4. Lovastatin did not improve spatial and temporal gait parameters in Ube3a^−/+^ mice. Several metrics of gait were assessed in Ube3a^−/+^ mice (AS) treated with lovastatin. Lovastatin at any dose did not have any effect on either (A) forelimb stance width or (D) hindlimb stance width in Ube3a^−/+^ mice. However, when treated with lovastatin, reductions in forelimb stride length were observed at 10 mg/kg or 100 mg/kg (B) and in the (E) hindlimb stride length at 10 mg/kg. Similarly, a dose of either 10 mg/kg or 100 mg/kg altered both the (C) forelimb stride frequency and (F) hindlimb stride frequency, exhibiting values closer to vehicle treated Ube3a^+/+^ mice (WT). (G) Reductions in forelimb stride duration were observed when treated with either 10 mg/kg or 100 mg/kg of lovastatin. (J) Hindlimb stride duration was also reduced to levels closer to Ube3a^+/+^ mice when treated with 10 mg/kg of lovastatin. Although no changes were seen at any dose in (H) forelimb propulsion duration, Ube3a^−/+^ animals treated with either 10 mg/kg or 100 mg/kg displayed reductions in (K) hindlimb propulsion duration. Finally, (I) forelimb stance duration was reduced when Ube3a^−/+^ mice were treated either 10 mg/kg or 100 mg/kg of lovastatin, and (L) hindlimb stance duration was reduced at each dose of lovastatin evaluated. Number of mice tested for gait: WT Veh N = 10, 10 mg/kg N = 8, 30 mg/kg N = 8, 100 mg/kg N = 7; AS Veh N = 9, 10 mg/kg N = 10, 30 mg/kg N = 9, 100 mg/kg N = 10. * p < 0.05, two-way ANOVA, one–way ANOVA followed by Dunnett’s post hoc analysis
For forelimb and hindlimb, stride duration, genotype and lovastatin dose effects were identified, using two-way ANOVAs (forelimb: Fig. 4G; F genotype(1, 67) = 50.81, p < 0.001; Fig. 4G; F dose inUbe3a^−/+^ (3, 67) = 2.956, p = 0.0304 and hindlimb: Fig. 4J; F genotype(1, 67) = 54.37, p < 0.0001; Fig. 4J; F dose inUbe3a^−/+^(3, 67) = 2.945, p < 0.0308). AS mice treated with 30 mg/kg of lovastatin displayed increased forelimb stride duration, analyzed with a one-way ANOVA and Dunnett’s post hoc analysis (30 mg/kg forelimb: p = 0.0124), compared to vehicle treated WT mice, but not versus the vehicle treated AS mice. AS mice treated with 30 mg/kg or 100 mg/kg of lovastatin displayed increased hindlimb stride duration, analyzed with a one-way ANOVA and Dunnett’s post hoc analysis (30 mg/kg, p < 0.0367 and 100 mg/kg, p < 0.0475), compared to vehicle treated WT mice, but not versus the vehicle treated AS mice. For forelimb and hindlimb stance duration, genotype effects were identified, using two-way ANOVAs (Fig. 4I; F genotype (1, 67) = 26.50, p < 0.001; Fig. 4L; F genotype (1, 67) = 26.88, p < 0.001). Lovastatin treatment at 30 mg/kg increased the forelimb stance duration in AS mice (Fig. 4I; F dose inUbe3a^−/+^(3, 43) = 2.560, p < 0.05), by a one-way ANOVA, followed by Dunnett’s post hoc analysis (30 mg/kg forelimb; p < 0.0417), compared to vehicle treated WT mice.
Hindlimb propulsion duration exhibited a genotype effect in the vehicle groups with increased duration in AS mice compared to WT mice, with a two-way ANOVA (Fig. 4K; F genotype (1, 67) = 35.10, p < 0.001). Lovastatin treatment at 30 mg/kg increased hindlimb propulsion duration, in AS mice (Fig. 4K; F dose inUbe3a^−/+^(3, 43) = 4.355, p < 0.005), by a one-way ANOVA, followed by Dunnett’s post hoc analysis (30 mg/kg forelimb: p < 0.0185), compared to vehicle treated WT mice.
Lovastatin disrupted novel object recognition in WT mice and did not improve novel object recognition in AS mice
Novel object recognition (NOR) was performed to evaluate improvement in cognition, resulting from lovastatin treatment. Vehicle treated WT mice spent more time investigating the novel object versus the familiar object, demonstrating intact recognition memory (Fig. 5A; t_(14)_ = 1.958, p = 0.0392), whereas vehicle treated AS mice did not (Fig. 5B; t_(16)_ = 0.8403, p = 0.4131), as previously published [18, 26]. Lovastatin treated WT mice disrupted these intact cognitive skills, and did not spend greater time investigating the novel object versus the familiar object, analyzed using within genotype paired t-tests (Fig. 5A; 10 mg/kg, t_(14)_ = 1.622, p = 0.1222; 30 mg/kg, t_(14)_ = 1.080, p = 0.2943; 100 mg/kg, t_(14)_ = 0.4734, p = 0.6438), or using discrimination index analyzed using one-way ANOVA (Fig. 5C; F dose inUbe3a^+/+^(3, 31) = 0.856, p = 0.474), nor by preference ratio analyzed using one-way ANOVA (Fig. 5D; F dose inUbe3a^+/+^(3, 31) = 0.856, p = 0.474), illustrating that lovastatin worsened the short term object recognition memory in WT mice, and has deleterious effects in typical control groups.Fig. 5. Lovastatin did not improve novel object recognition in Ube3a^−/+^ and disrupted novel object recognition in Ube3a^+/+^ mice. A In the novel object recognition task, vehicle treated Ube3a^+/+^ mice investigated the novel object for significantly longer than the familiar object, as expected. * p < 0.05, t-test of novel versus familiar. When treating Ube3a^+/+^ mice with any dose of lovastatin, the investigation time of the novel object was reduced, and no preference for investigating the novel object was observed. B Ube3a^−/+^ mice showed no preference for investigating the novel object when treated with vehicle or any dose of lovastatin. * p < 0.05, t-test of novel versus familiar object investigation. C Using discrimination index to measure the varying doses of lovastatin on Ube3^+/+^ and Ube3a^−/+^ mice, no significant cognitive effect was observed, via one-way ANOVA, followed by Dunnett’s posthoc comparisons. D Using preference ratio to measure the varying doses of lovastatin on Ube3a^+/+^ and Ube3a^−/+^ mice, no significant cognitive effect was observed, via one-way ANOVA, followed by Dunnett’s post hoc comparisons
Lovastatin did not improve novel object recognition in the AS mice, illustrated by the fact that AS mice did not spend more time investigating the novel object versus the familiar object following lovastatin treatment, analyzed using within genotype paired t-tests (Fig. 5B; 10 mg/kg, t_(16)_ = 0.8403, p = 0.4037; 30 mg/kg, t_(16)_ = 1.181, p = 0.2573; 100 mg/kg, t_(14)_ = 0.8720, p = 0.4037), or using discrimination index analyzed using one-way ANOVA (Fig. 5C; F dose inUbe3a^−/+^(3, 28) = 3.163, p = 0.8135), nor by preference ratio analyzed using one-way ANOVA (Fig. 5D; F dose inUbe3a^−/+^(3, 28) = 3.163, p = 0.8135).
To rule out artifactual influence of motor activity, and behavioral changes resulting from weight differences, all mice were weighed throughout behavioral testing. Vehicle treated WT and AS mice did not exhibit significantly different weights during testing (Fig. S1 A), and no dose of lovastatin influenced weight in either subject group of WT or AS mice (Figs. S1B, S1C).
Since novel object recognition heavily relies on intact motor ability, to explore and investigate the properties of objects, the observed sedative effects in the open field, following lovastatin in the WT mice, limits the ability to make strong conclusions, regarding the cognitive aspect of novel object recognition, or the cognitive effect of lovastatin, from the adverse sedating effects of lovastatin, without additional cognitive analysis. Unfortunately, our secondary assay, spontaneous alternation in the Y-maze, is also heavily dependent on motor ability; yet more importantly, no genotype effect was observed between WT and AS mice (Figure S2). Since AS did not have a Y-maze deficit, lovastatin’s potential improvement in spontaneous alternation, as an index of cognitive performance, could not be measured via the Y-maze (Figure S2).
Discussion
There is a large volume of preclinical and clinical knowledge around small molecule therapeutics which can be repurposed to treat the most debilitating domains of a variety of neurodevelopmental disorders (NDDs), including Angelman Syndrome (AS). For example, cannabidiol, neurosteroids, lovastatin, folic acid and betaine have been shown efficacious for seizure control in AS [37, 64, 90–92]. Despite the ease with which repurposed compounds can be evaluated in AS, non-Ube3a based therapeutic intervention has been far less studied than gene-targeted replacement-based therapies. With a vast number of compounds available and proven to be safe and well tolerated, assessing repurposed compounds, especially those with mechanisms of action up or down stream to the ubiquitination pathway, remains an unmet need.
For the first time, we report unexpectedly detrimental effects of lovastatin in WT subject mice during behavioral assays of motor abilities, including the total and horizontal activity assessments and when total activity over 30-min is summed. Furthermore, lovastatin reduced summed vertical activity in subject WT mice following 30 mg/kg and 100 mg/kg and AS subject mice treated with 10 mg/kg, 30 mg/kg and 100 mg/kg. The corroborating motor assay of motor coordination and motor learning, the accelerating rotarod, illustrated the well reported impairment in AS mice [26, 27, 44, 65, 91, 93, 94] which has been published repeatedly by us and others, while no effect of lovastatin was observed on the rotarod in either genotype.
We also discovered that numerous gait metrics that are impaired in AS, are impaired in WT mice at the 100 mg/kg dose of lovastatin. The nine sub-indices that differ between AS and WT were not fully restored by lovastatin administration across a variety of doses. The forelimb and hindlimb stride length were extended in vehicle treated AS mice and 30 mg/kg of lovastatin in AS mice caused this group to statistically differ from WT vehicle mice in forelimb stride length (Fig. 4B), while 30 mg/kg and 100 mg/kg of lovastatin in AS mice caused this group to statistically differ from WT vehicle mice in hindlimb stride length (Fig. 4E). Further, the forelimb and hindlimb stride frequency that were reduced in vehicle treated AS mice (Fig. 4C, F), were further reduced by 30 mg/kg of lovastatin in AS mice in forelimb stride frequency and 30 mg/kg and 100 mg/kg in hindlimb frequency (Fig. 4C, F). Finally, increased forelimb and hindlimb stride duration were detected in vehicle treated AS mice (Fig. 4I, L). 30 mg/kg of lovastatin in AS mice also increased forelimb stride duration and both 30 mg/kg and 100 mg/kg increased hindlimb stride duration, yet none of these alterations improved AS gait parameters to WT levels (Fig. 4G, J). AS propulsion duration did not differ in the forelimbs but the hindlimb propulsion duration was elevated, compared to WT. 30 mg/kg of lovastatin treatment in AS mice resulted in a reduction of the elevated propulsion duration, but not to a level that brought AS mouse gait to WT levels. Finally, stance duration was elevated in AS mice in both fore and hindlimb assessments, compared to WT. While 30 mg/kg of lovastatin in AS mice lowered the elevated stance duration in the forelimb index, it was not a large enough of an effect size to normalize AS stance duration to WT levels. It is worth noting a few mild gait improvements in AS mice may be attributed to lovastatin treatment but given that the effect size was so small that differences could not be observed compared to AS vehicle, any concrete interpretations are not able to be reached. Further, given the observed sedating effects of lovastatin in Fig. 1, any gait improvements on parameters that exhibited AS increases, could be confounded by the sedating effects of lovastatin.
Therefore, the preponderance of the data suggests that 100 mg/kg of lovastatin worsened gait in WT mice and that any improvements to gait in AS mice from lovastatin treatment, were either of subtle small effect sizes or may be confounded by sedation. Yet, the gait findings regarding lovastatin treatment in AS were not completely null, and our work illustrated a few subtle effects over more convincing sizeable effects.
There were no significant differences in the forelimb stance width, forelimb stride frequency, or hindlimb stride frequency in WT mice, when treated with vehicle or any dose of lovastatin (Fig. 3A, C, F). Our tailored focus on motor abilities, such as exploration, coordination and gait sharpened our observations of lovastatin, mildly rescuing some metrics with 30 mg/kg and 100 mg/kg doses of lovastatin. Rescue was demonstrated by numerous AS altered metrics, brought closer to WT levels. Stride length and frequency are two metrics that we have previously reported to be altered in AS mouse models and individuals with AS [26, 47, 87–89]. When compared to WT mice, we observed increases in stride length, stride duration, stance duration, and propulsion duration alongside reduced stride frequency in AS mice, previously observed in our lab [18, 26, 47]. Taken together, these metrics are indicative of longer, slower steps, which mimics what is seen in individuals with AS [87, 89, 95]. Stride length in the AS model averages 6–6.5 cm, with a stride frequency of approximately 3.2–3.6 steps/s, versus WT controls that exhibit stride lengths of approximately 5.2–6.0 cm and stride frequencies of 3.8–4.2 steps/s. Both 10 mg/kg and 100 mg/kg of lovastatin treatment in AS mice resulted in reduced stride lengths with an average of 5.8 cm and increased stride frequencies of approximately 4.0 steps/s, returning their values closer to WT. Y-axis values in this study reproduced earlier research using the AS mouse model [26, 47].
Gait analysis using an automated treadmill was performed using the DigiGait™ system. Quantitative gait assessments are objective, translational tools, for which automated equipment exists to acquire spatial and temporal indices in rodent models of neurodevelopmental disorders (NDDs). Similar equipment and data are being utilized, collected, and reported for clinical populations, suggesting that gait is increasing in popularity as a useful quantitative, objective outcome measure, with biomarker potential. Preclinical data have been validated by multiple laboratories, assessed across a developmental trajectory, and possess reliability and rigor by the number of metrics captured and robustness, as the findings are not specific to a piece or brand of equipment. To date, gait reports have provided preclinical data that is similar to systems for measuring human gait that utilize pressure sensitive walkways, instrumented gait analysis, and activity monitoring devices, in genetic NDDs, such as Williams, Phelan McDermid and Angelman Syndromes, as well as Neurofibromitosis-1 [47, 96–99].
Angelman Syndrome (AS) is a rare genetic disorder characterized by intellectual disabilities, motor deficits, impaired communication, and an autism spectrum disorder diagnosis. Movement disorders affect every individual with AS, regardless of the specific molecular causal subtype of the disorder. Motor impairments are the most prevalent and adverse impairment of the complex AS symptomology, which also includes recurring seizures [90, 100, 101]. Examples include spasticity, ataxia of gait (observed in most ambulatory individuals), tremors, and muscle weakness [87]. Motor behavior is highly conserved across species, exemplified by gene expression and neural circuitry, highlighting their utility as a translational outcome.
Motor deficits have been key in the study of mouse models of AS, as dysfunction on the rotarod and reduced activity have been the most consistently reported behavioral phenotypes. Clinically, motor delays and ataxia have been observed over time, and individuals with AS develop altered gait, which may signal a progressive decline in mobility. This decline is associated with decreased endurance, a more sedentary lifestyle, and diminished quality of life. Gait measurements offer a potential indicator of therapies, at a time when NDD clinical trials often lack appropriate outcomes measures. Moreover, interventions that improve gait may “spark” improvements in additional behavioral domains, offering the potential to prevent and avoid progressive declines in endurance and activity across the lifespan of those with AS [89]. This manuscript illustrates and reproduces AS mouse model deficits in overall motor ability to navigate an open field, motor coordination dysfunction on the rotarod, and gait using the automated DigiGait treadmill. However, any clear positive influence of lovastatin treatment on gait in AS mice was not evident, and the adverse influence of lovastatin treatment in WT and AS mice was observed.
Although there are no drug therapies that address the underlying causes of AS, new molecular therapies that are targeted to specific proteins, RNA, or DNA hold tremendous promise for the future. Although this strategy is promising, off-target effects and an unknown safety profile may extend the time it takes for these and other gene editing therapies to reach FDA approval and clinical use. Another promising therapeutic candidate was topotecan which reactivated the paternal allele utilizing Ube3a^+/YFP^ mice by inhibiting transcriptional progression of the paternal ATS [2, 65, 102, 103]. When injected unilaterally into the ventricle of mice with paternal Ube3a-YFP, yellow immunofluorescence from paternal expression was observed in the treated hemisphere lasting up to 12 weeks. Although topotecan was approved by the FDA for various cancers including ovarian and lung cancer [65], its lack of specificity and general toxicity [103] has limited advancement of the drug to the clinic for AS. Moreover, the intracerebroventricular route of administration is not desirable for translational pursuit, unless no other possibilities exist. Thus, while topotecan is not a viable treatment for AS, this research paved the road for future studies focusing on paternal reactivation and its mechanistic class, topoisomerase inhibitors [103], and more broadly for repurposing and small molecule therapeutics for AS.
In a preclinical study, lovastatin protected Ube3a^−/+^ mice from audiogenic seizures and reduced long burst firing in slices from these mice [37]. This suggests that there could be an anticonvulsant effect of lovastatin in AS mice. A related compound, simvastatin, also improved cognitive and social function in an AS mouse model. The authors related this effect to mechanistically restoring HDAC1/2 activity in these mice [104]. Without further evaluation, it is difficult to predict the precise effect that lovastatin would have in humans with AS, and it is possible that different routes of administration, post-treatment intervals, or dosing schemes might improve efficacy.
Lovastatin was well-tolerated in multiple clinical trials treating children with Neurofibromatosis Type 1 and some verbal and non-verbal memory improvements were observed [105, 106], however, a larger placebo-controlled trial showed no improvement in cognitive ability [107]. Clinical trials treating Fragile X Syndrome with lovastatin showed similar safety and tolerability at multiple doses and promising improvements in cognition and Aberrant Behavioral Checklist–Community (ABC-C) scores [108, 109]. Despite that, no clinical study has been conducted specifically for AS patients, the safety and efficacy results from trials of other NDDs with similar affected domains, offered promising potential for repurposing lovastatin for the treatment of AS.
In AS patients, motor skills can limit normal functions such as self-feeding, verbal ability, and cognition skills. Even if the understanding and intent is present at the communicative level, the AS patient may be unable to physically execute normal functions due to motor impairments, thus highlighting the fact that motor skills are a critical limiting factor in basic functions and any minimal improvement is highly desirable. Currently, it is our goal to identify specific parameters being used in AS clinics to identify distinct motoric and gait signatures in the Ube3a^−/+^ mice and rats, and identify therapeutics that improve these impairments, regardless of class or mechanism of action.
In summary, we and others [96, 97, 99] have been investigating the translatability of gait and find it to be a useful, objective, translational, cross species biomarker for preclinical models of genetic NDDs and for assessing the onset of impairments and/or progressive declines/regression in rodent models. Of additional interest in this report were the adverse effects we reported in the Ube3a^+/+^ mice resulting from lovastatin treatment in the open field, spatial and temporal metrics of gait and in the novel objection recognition and Y-maze learning and memory assays. Given the translatable nature of motor activity and gait analysis, the effects, that we observed when treating Ube3a^+/+^ and Ube3a^−/+^ mice with lovastatin may have clinical relevance.
Supplementary Information
Supplementary Material 1.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Meng L, Person RE, Huang W, Zhu PJ, Costa-Mattioli M, Beaudet AL. Truncation of Ube 3a-ATS unsilences paternal Ube 3a and ameliorates behavioral defects in the Angelman syndrome mouse model. P Lo S Genet. 2013;9(12). 10.1371/journal.pgen.1004039. Epub 20131226. Pub Med PMID: 24385930; PMCID: PMC 3873245.10.1371/journal.pgen.1004039 PMC 387324524385930 · doi ↗ · pubmed ↗
- 2Ottenhoff MJ, Krab LC, Elgersma Y. Considerations for Clinical Therapeutic Development of Statins for Neurodevelopmental Disorders. e Neuro. 2020;7(2)ENEURO.0392-19.2020. 10.1523/ENEURO.0392-19.2020. Epub 20200306. Pub Med PMID: 32071072; PMCID: PMC 7070444.10.1523/ENEURO.0392-19.2020 PMC 707044432071072 · doi ↗ · pubmed ↗
- 3Muscas M, Seo SS, Louros SR, Osterweil EK. A Differential Effect of Lovastatin versus Simvastatin in Neurodevelopmental Disorders. e Neuro. 2020;7(4):ENEURO.0162-20.2020. 10.1523/ENEURO.0162-20.2020. Epub 20200813. Pub Med PMID: 32651266; PMCID: PMC 7433894.10.1523/ENEURO.0162-20.2020 PMC 743389432651266 · doi ↗ · pubmed ↗
- 4Muscas M, Louros SR, Osterweil EK. Lovastatin, not Simvastatin, Corrects Core Phenotypes in the Fragile X Mouse Model. e Neuro. 2019;6(3):ENEURO.0097-19.2019. 10.1523/ENEURO.0097-19.2019. Epub 20190612. Pub Med PMID: 31147392; PMCID: PMC 6565377.10.1523/ENEURO.0097-19.2019 PMC 656537731147392 · doi ↗ · pubmed ↗
- 5Berg EL, Pride MC, Petkova SP, Lee RD, Copping NA, Shen Y, Adhikari A, Fenton TA, Pedersen LR, Noakes LS, Nieman BJ, Lerch JP, Harris S, Born HA, Peters MM, Deng P, Cameron DL, Fink KD, Beitnere U, O'Geen H, Anderson AE, Dindot SV, Nash KR, Weeber EJ, Wohr M, Ellegood J, Segal DJ, Silverman JL. Translational outcomes in a full gene deletion of ubiquitin protein ligase E 3A rat model of Angelman syndrome. Transl Psychiatry. 2020;10(1):39. Epub 2020/02/19. 10.1038/s 41398-020-0720-2. Pub Med PMID: 32 · doi ↗ · pubmed ↗
- 6Born HA, Martinez LA, Levine AT, Harris SE, Mehra S, Lee WL, Dindot SV, Nash KR, Silverman JL, Segal DJ, Weeber EJ, Anderson AE. Early Developmental EEG and Seizure Phenotypes in a Full Gene Deletion of Ubiquitin Protein Ligase E 3A Rat Model of Angelman Syndrome. e Neuro. 2021;8(2). Epub 20210324. 10.1523/ENEURO.0345-20.2020. Pub Med PMID: 33531368; PMCID: PMC 8114899.10.1523/ENEURO.0345-20.2020 PMC 811489933531368 · doi ↗ · pubmed ↗
- 7Copping NA, Silverman JL. Abnormal electrophysiological phenotypes and sleep deficits in a mouse model of Angelman Syndrome. Mol Autism. 2021;12(1):9. Epub 2021/02/08. 10.1186/s 13229-021-00416-y. Pub Med PMID: 33549123; PMCID: PMC 7866697.10.1186/s 13229-021-00416-y PMC 786669733549123 · doi ↗ · pubmed ↗
- 8Petkova SP, Adhikari A, Berg EL, Fenton TA, Duis J, Silverman JL. Gait as a quantitative translational outcome measure in Angelman syndrome. Autism Res. 2022;15(5):821–33. Epub 20220310. 10.1002/aur.2697. Pub Med PMID: 35274462; PMCID: PMC 9311146.10.1002/aur.2697 PMC 931114635274462 · doi ↗ · pubmed ↗