U2af1S34F and U2af1Q157R myeloid neoplasm-associated hotspot mutations induce distinct hematopoietic phenotypes in mice
Michael O. Alberti, Sridhar Nonavinkere Srivatsan, Jin Shao, Dennis L. Fei, Mengou Zhu, Clauida Cabrera Pastrana, Sarah Grieb, Timothy A. Graubert, Omar Abdel-Wahab, Matthew J. Walter

TL;DR
Two mutations in the U2AF1 gene cause different blood-related issues in mice, suggesting they may affect disease differently in humans.
Contribution
The study shows that U2AF1S34F and U2AF1Q157R mutations lead to distinct hematopoietic and splicing effects in mice.
Findings
U2af1S34F causes more severe blood and bone marrow cell count reductions than U2af1Q157R.
The two mutations affect different target genes and splicing patterns in mice and humans.
The mutations co-occur with different gene mutations in patients, indicating varied disease mechanisms.
Abstract
Recurrent somatic mutations in the spliceosome genes SF3B1, SRSF2, and U2AF1 are frequently identified in patients with myeloid neoplasms, such as myelodysplastic syndromes. We characterized the in vivo consequences of expressing two hotspot mutations in U2AF1 that code for the S34F and Q157R substitutions. Our results indicate that the two mutations induce distinct hematopoietic phenotypes in mice, suggesting that the U2AF1S34F and U2AF1Q157R mutations should not be conflated as they may impact disease pathogenesis differently in patients. Mice expressing U2af1S34F have a more severe reduction in their blood and bone marrow cell counts and reduced stem cell repopulating ability, compared to mice expressing U2af1Q157R. The expression and splicing of target genes are largely unique between the mutations, in both mouse and human samples, potentially driving the phenotypic differences…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
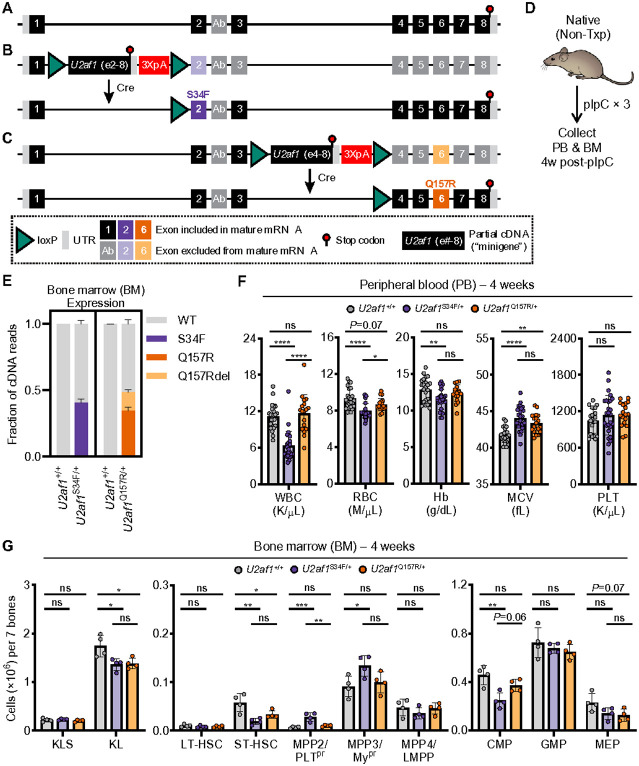 Figure 1
Figure 1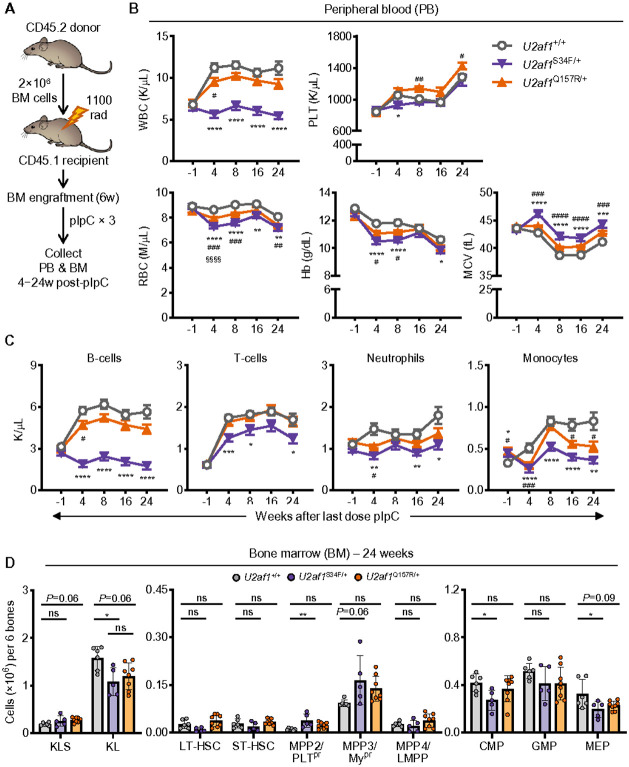 Figure 2
Figure 2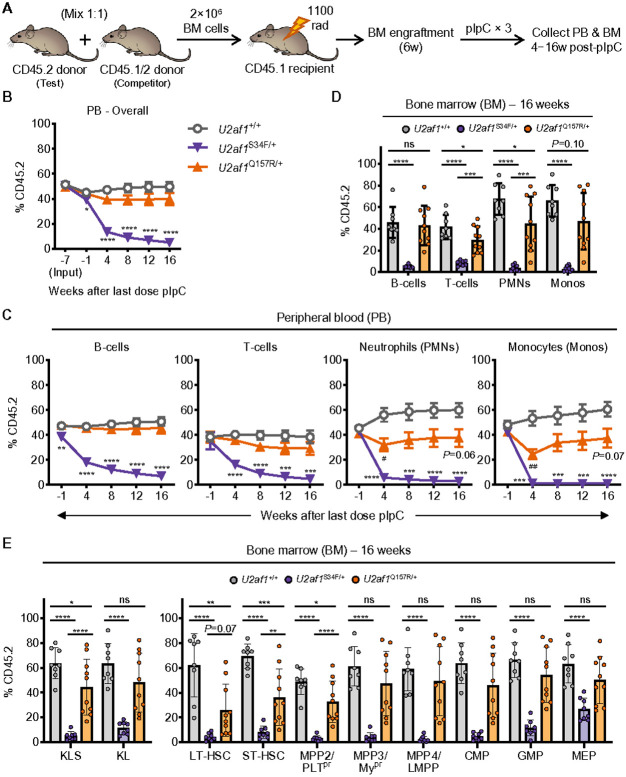 Figure 3
Figure 3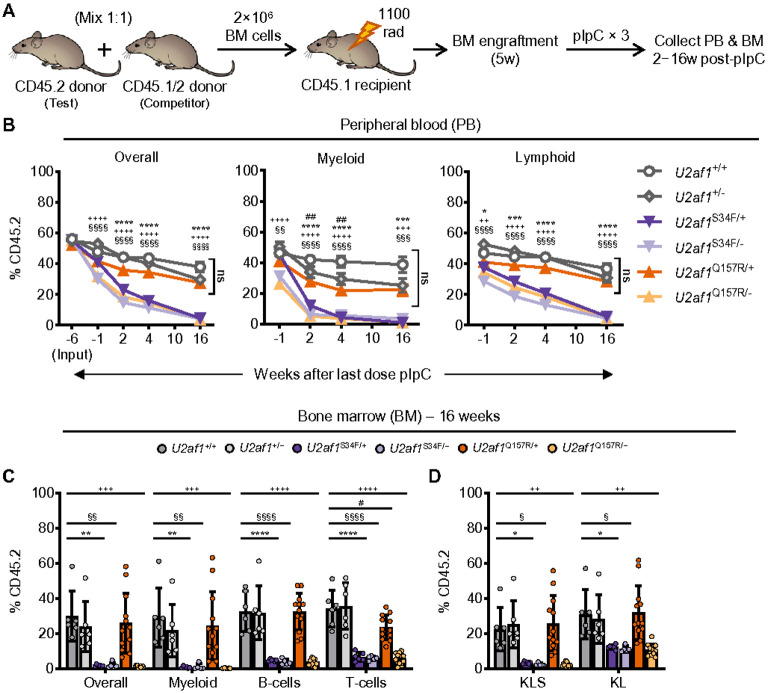 Figure 4
Figure 4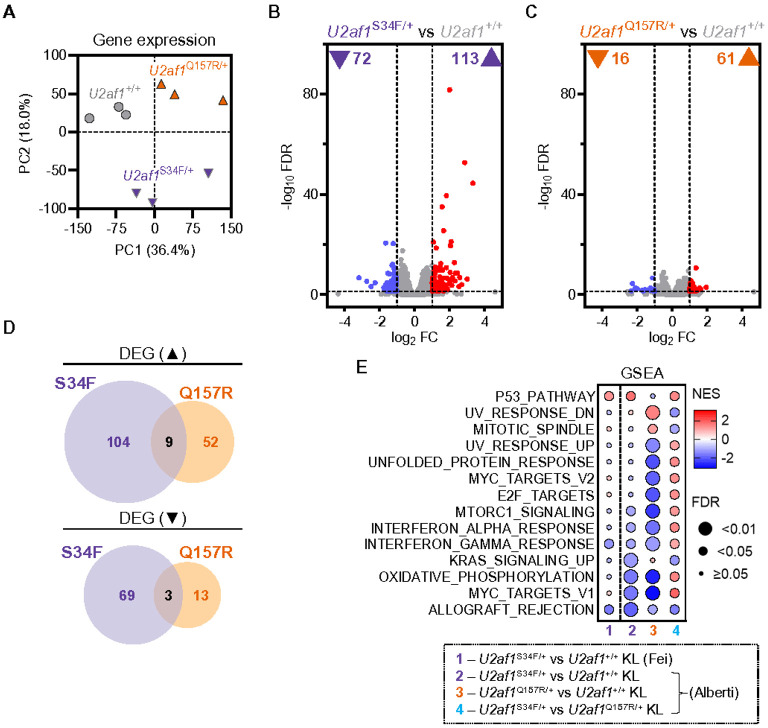 Figure 5
Figure 5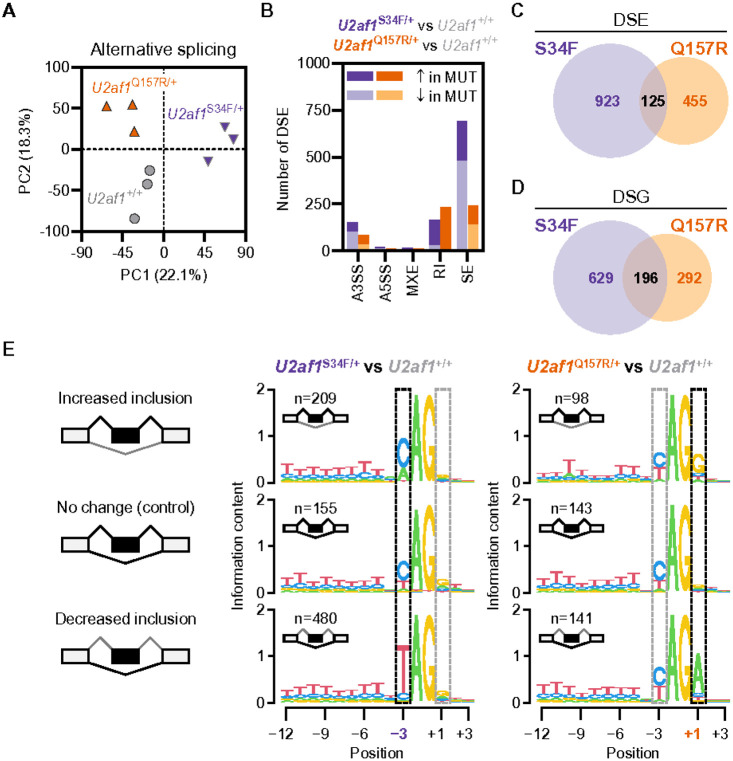 Figure 6
Figure 6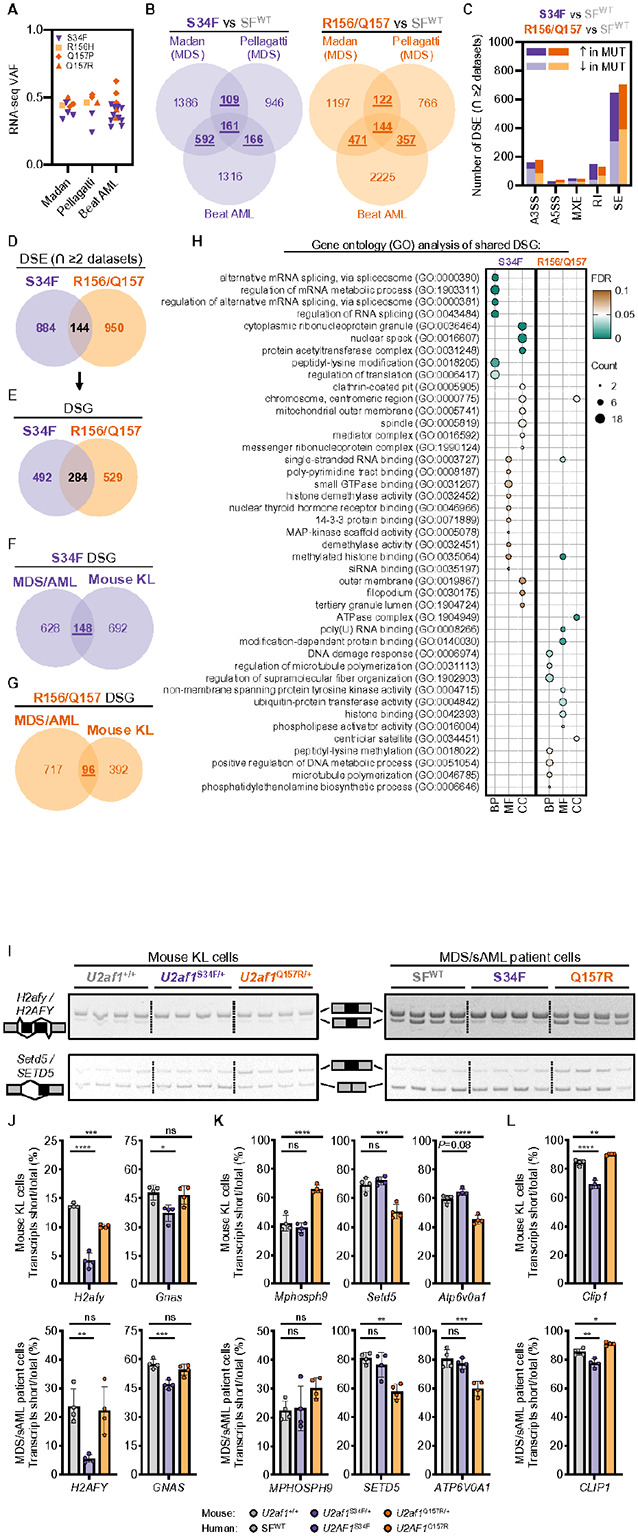 Figure 7
Figure 7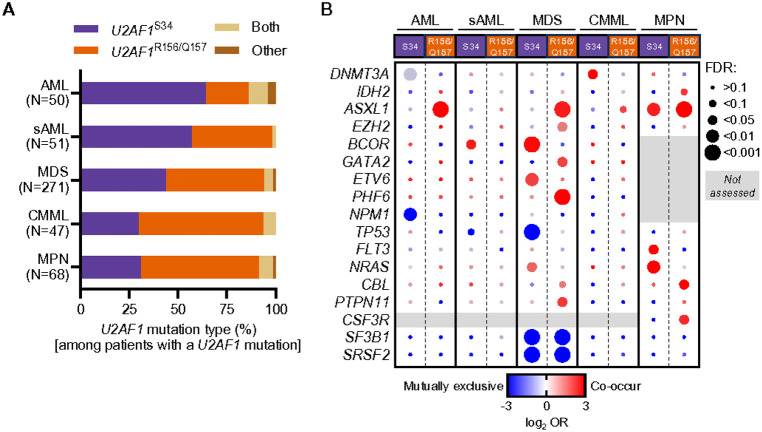 Figure 8
Figure 8Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsAcute Myeloid Leukemia Research · Lymphoma Diagnosis and Treatment · Myeloproliferative Neoplasms: Diagnosis and Treatment
INTRODUCTION
Recurrent somatic mutations in a subset of spliceosome genes (SF3B1, SRSF2, and U2AF1) are frequently identified (30–60% depending on disease phenotype) in patients afflicted with myelodysplastic syndromes (MDS), myeloproliferative neoplasms (MPN) such as myelofibrosis (MF), MDS/MPN overlap disorders such as chronic myelomonocytic leukemia (CMML), and secondary acute myeloid leukemia (sAML).^1-8^ These heterozygous and mutually exclusive mutations are enriched in hotspot codons in these 3’ splicing factor proteins resulting in aberrant alternative mRNA splicing in hematopoietic cells. However, each mutant protein predominantly affects a distinct set of alternatively spliced downstream target genes suggesting that common downstream pathway alterations or cellular response to mutation expression, rather than specific shared splicing targets, may be responsible for MDS phenotypes, including dysplasia, ineffective hematopoiesis, and cytopenias.^9-11^
U2AF1 provides a unique opportunity to address this question because it has two hotspot positions (serine 34 [S34] and glutamine 157 [Q157]) that are each commonly mutated in MDS and are associated with unique mRNA splicing consequences.^12^ In addition, U2AF1 S34 and Q157 codon mutations co-occur with mutations in different genes (e.g., BCOR and ASXL1, respectively) and patients with these mutations may have different hematopoietic phenotypes—highlighting that these mutations may induce distinct phenotypes.^13-16^ We asked if the splicing differences resulting from S34F and Q157R mutations were thus associated with different or similar effects on hematopoiesis. To do so, we characterized and compared an established conditional S34F knock-in mouse model^17^ to a new Cre/lox conditional mouse model with the Q157R mutation knocked-in to the endogenous U2af1 locus, in order to directly study the hematopoietic phenotype, transcriptional, and mRNA splicing consequences of individual U2AF1 gene mutations in vivo.
MATERIALS AND METHODS
Animal models and experimental details
Experiments were performed per institutional guidelines for care and use of laboratory animals and approved by the Institutional Animal Care and Use Committee of Washington University in St. Louis (WUSTL). U2af1^Q157R/+^ (MiniGene Q157R or ‘MGQ157R’) conditional knock-in mice were generated by Biocytogen (Waltham, MA). A full description of the targeting construct is described in the Supplementary Methods. U2af1^S34f/+^ (‘MGS34F’) conditional knock-in (Jackson Laboratory [JAX] Stock #032638, Bar Harbor, ME),^17^ U2af1^fl/+^ conditional knockout (JAX Stock #037015),^18^ and Mx1-Cre (JAX Stock #003556)^19^ mice are described elsewhere. B6.SJL-Ptprc^a^Pepc^b^/BoyCrCrl or ‘CD45.1’ recipient mice were purchased from Charles River Laboratories (Stock #564). Heterozygous CD45.1/CD45.2 mice were bred by crossing C57BL/6J (B6; JAX Stock #000664) to B6.SJL-Ptprc^a^Pepc^b^/BoyJ (JAX Stock #002014). All mouse lines were on a B6 background. Genotyping primers are listed in Supplementary Table 1.
Details pertaining to bone marrow (BM) transplant, peripheral blood (PB) sampling and analysis, and flow cytometry setup and population gating are described in the Supplementary Methods.
mRNA-sequencing (RNA-seq) and bioinformatics
BM myeloid progenitor (c-kit^+^Lineage^−^Sca-1^−^; KL) cells were sorted into FACS buffer and gDNA-depleted total RNA were purified from cell pellets using the NucleoSpin RNA Plus XS Micro Kit (Macherey-Nagel, Allentown, PA) in RNase-free water. RNA concentration and RIN were measured by Bioanalyzer (Agilent, Santa Clara, CA) and then cDNA libraries for RNA-seq were prepared by KAPA RNA HyperPrep Kit with RiboErase (Cat #KK8560/61; Roche, Indianapolis, IN). Detailed library preparation and bioinformatic analyses are described in Supplementary Methods.
Details pertaining to the bioinformatics and reanalysis of published MDS and AML RNA-seq datasets, analysis of U2AF1 hotspot mutation co-occurrence in myeloid malignancies, and confirmation of splicing changes in mouse KL cell and MDS/sAML patient samples are also described in the Supplementary Methods. All patients provided written consent on a protocol approved by the WUSTL Human Studies Committee. Clinical characteristics of patients who donated research samples are listed in Supplementary Table 2.
Statistics
Data were analyzed and visualized using GraphPad Prism 10 software (Boston, MA). Statistical tests are described in each figure legend. P < 0.05 was considered statistically significant.
RESULTS
Establishing a mouse model with conditional knock-in of the Q157R mutation at the U2af1 locus.
A conditional (Cre/lox-mediated) knock-in of the S34F mutation at the U2af1 locus (MGS34F or U2af1^S34F/+^) was previously generated (Fig. 1A,B and Supplementary Fig. 1A).^17^ To allow for direct comparison with the MGS34F mouse, a similar strategy was used to generate a conditional (Cre/lox-mediated) Q157R mutant allele at the endogenous U2af1 locus (MGQ157R or U2af1^Q157R/+^) of B6 mice (Fig. 1C and Supplementary Fig. 1B). Successful introduction of the targeting vector at the U2af1 locus was confirmed by Southern blot and Sanger sequencing (Supplementary Fig. 1C). To confirm Cre/lox-mediated hematopoietic expression of U2af1^Q157R^ mRNA and assess the short-term effects of U2AF1^Q157R^ in a non-transplant model (i.e., native hematopoiesis), we crossed heterozygous U2af1^Q157R/+^ mice to Mx1-Cre transgenic mice (Fig. 1D-G and Supplementary Fig. 1D-I). Mx1-Cre is expressed in hematopoietic lineage cells following administration of polyinosinic-polycytidylic acid (pIpC).^19^
Four weeks after pIpC treatment of U2af1^Q157R/+^;Mx1-Cre mice, the U2af1 wild-type (WT) and Q157R alleles were expressed at similar levels in BM myeloid progenitor (KL) cells by targeted NGS amplicon sequencing of cDNA (Fig. 1E). As expected, the WT and S34F alleles were also expressed at similar levels in BM KL cells from U2af1^S34F/+^;Mx1-Cre mice and only the WT allele was detected in U2af1^+/+^;Mx7-Cre control mice (Fig. 1E). It was previously reported that the Q157R mutation in U2AF1 creates an alternative 5’ splice site that leads to expression of a minor U2AF1 isoform (termed ‘Q157Rdel’) with in-frame deletion of four amino acids immediately following the Q157R mutant codon. The U2af1^Q157R/+^ mouse model recapitulates expression of the Q157Rdel isoform in BM KL cells (Fig. 1E and Supplementary Fig. 1D).
U2AF1 S34F and U2AF1Q157R cause different hematopoietic changes in mice.
To determine if S34F and Q157R result in similar short-term effects on native hematopoiesis, we performed complete blood counts and flow cytometric analysis on PB samples from U2af1^Q157R/+^, U2af1^S34F/+^, and U2af1^+/+^ control mice (all Mx1-Cre^+^) four weeks after pIpC treatment. Consistent with previous characterization,^17^ U2af1^S34F/+^ mice had no change in platelet counts, modestly reduced red blood cell (RBC) counts and hemoglobin levels (with elevated mean corpuscular volume [MCV]), and markedly reduced white blood cell counts compared to U2af1^+/+^ mice (Fig. 1F). Flow cytometric analysis of U2af1^S34F/+^ PB and BM demonstrated significant reductions in both myeloid and lymphoid lineages (Supplementary Fig. 1E,F). In contrast, U2af1^Q157R/+^ mice had no significant PB or BM changes except for elevated MCV (Fig. 1F and Supplementary Fig. 1E,F). Assessment of BM hematopoietic stem and progenitor cells (HSPC) four weeks after pIpC treatment revealed that U2af1^S34F/+^ mice had significantly reduced numbers of short-term hematopoietic stem cell (ST-HSC), KL, and common myeloid progenitor (CMP) populations with increased numbers of multipotent progenitor (MPP)2 and MPP3 populations compared with control mice (Fig. 1G). U2af1^Q157R/+^ mice also had significantly reduced numbers of ST-HSC and KL populations and non-significant reductions in both CMP and megakaryocyte-erythroid progenitor (MEP) cells compared with control mice (Fig. 1G). U2af1^S34F/+^ mice had a significant block in erythroid development in the BM and spleen, with an increased proportion of immunophenotypically defined nucleated erythroblasts (Ter119^lo/hi^CD71^hi^) and a decreased proportion of enucleated erythrocytes (Ter119^hi^CD71^lo^). In contrast, U2af1^Q157R/+^ mice had a smaller but non-significant, increase in Ter119^hi^CD71^hi^ cells in the spleen (Supplementary Fig. 1G-I).
To better evaluate the cell-intrinsic effects of both mutants on hematopoiesis, we transplanted BM from U2af1^Q157R/+^, U2af1^S34F/+^, or U2af1^+/+^ control mice (CD45.2^+^; all Mx1-Cre^+^) into lethally irradiated WT congenic recipient mice (CD45.1^+^). Following engraftment, we treated mice (including controls) with pIpC to induce expression of S34F and Q157R in donor-derived cells (Fig. 2A). Four weeks after pIpC treatment, PB (Fig. 2B,C) and BM changes (Supplementary Fig. 2A,C) reflected similar overall trends observed in native hematopoiesis (Fig. 1F and Supplementary Fig. 1E) for both mutant mice. At 24 weeks, both mutant mice had significantly reduced PB RBC counts with increased MCV, as well as decreased hemoglobin in U2af1^S34F/+^ mice. U2af1^Q157R/+^ mice also had mildly increased platelet counts (Fig. 2B). U2af1^S34F/+^ mice had significantly reduced PB and BM myeloid and lymphoid lineage cells, while U2af1^Q157R/+^ mice had significantly decreased PB monocytes and a non-significant increase in BM monocytes (Fig. 2C and Supplementary Fig. 2D). Although myeloid and lymphoid lineages were significantly decreased in U2af1^S34F/+^ mouse spleens at 4 weeks, there were no significant changes at 24 weeks (Supplementary Fig. 2B,E). HSPC populations reflected similar significant overall trends at 24 weeks compared to 4 weeks for U2af1^S34F/+^ mice (Fig. 2D and Supplementary Fig. 2C). U2af1^Q157R/+^ mice also showed similar, but non-significant, trends in HSPC population numbers at 24 weeks compared to 4 weeks (Fig. 2D and Supplementary Fig. 2C).
U2af1 S34F/+ HSCs are significantly more impaired than U2af1Q157R/+ HSCs in BM repopulation assays.
To compare the effects of S34F or Q157R expression on HSC reconstitution capacity, we performed competitive BM transplantation experiments. Lethally irradiated WT congenic recipient mice (CD45.1^+^) were transplanted with whole BM ‘test’ cells from U2af1^Q157R/+^, U2af1^S34F/+^, or U2af1^+/+^ control mice (CD45.2^+^; all Mx1-Cre^+^) mixed with an equal number of competitor BM cells from WT congenic mice (CD45.1^+^/CD45.2^+^). Following engraftment, we treated mice (including controls) with pIpC to induce expression of S34F and Q157R in donor-derived cells (Fig. 3A). Consistent with previous characterization,^17^ we observed significant multi-lineage reductions in PB, BM, and spleen donor cell chimerism (CD45.2^+^) for U2af1^S34F/+^ compared to U2af1^+/+^ test cells (Fig. 3B-D and Supplementary Fig. 3). In contrast, the reduction in overall and multilineage PB donor cell chimerism for U2af1^Q157R/+^ compared to U2af1^+/+^ test cells was less severe relative to U2af1^S34F/+^ test cells (Fig. 3B,C). In addition, there were variable reductions in donor cell chimerism of PB, BM, and spleen myeloid lineages for U2af1^Q157R/+^ compared to U2af1^+/+^ test cells (Fig. 3C,D and Supplementary Fig. 3). Donor cell chimerism for all BM HSPC populations were significantly reduced for U2af1^S34F/+^ compared to U2af1^+/+^ test cells (Fig. 3E). However, reduced U2af1^Q157R/+^ donor cell chimerism was restricted to the HSC and MPP2 populations, but not to the same degree as for U2af1^S34F/+^ (Fig. 3E).
Hemizygous U2af1Q157R/− and U2af1S34F/− HSCs are both severely impaired in BM repopulation assays.
We previously demonstrated that cell survival and reconstitution capacity are severely reduced for HSCs that express mutant U2AF1^S34F^ without WT U2AF1 expression (hemizygous U2af1^S34F/−^).^18^ Given the mild reconstitution defect observed for U2af1^Q157R/+^ cells, we hypothesized that mutant U2AF1^Q157R^ cells may not require the expression of WT U2AF1 for cell survival. To test this, we performed competitive BM transplantation experiments using test cells from three additional genotypes of mice: U2af1^Q157R/−^, U2af1^S34F/−^, and U2af1^+/−^ mice (all Mx1-Cre^+^; Fig. 4A). Hemizygous conditional knock-in mice were generated by crossing heterozygous floxed mutant (S34F or Q157R) mice to heterozygous floxed knockout mice. Consistent with previous characterization,^18^ we noted a rapid and significant loss in mature cell and HSPC donor cell chimerism (CD45.2^+^) in the PB, BM, and spleen for hemizygous U2af1^S34F/−^ (but not U2af1^+/−^) compared to U2af1^+/+^ test cells following administration of pIpC (Fig. 4B-D and Supplementary Fig. 4A,B). We also observed an identical rapid loss in mature cell and HSPC chimerism for hemizygous U2af1^Q157R/−^ compared to U2af1^+/+^ test cells (Fig. 4B-D and Supplementary Fig. 4A,B). This indicates that the expression of WT U2AF1 is required for the viability of either U2AF1^S34F^ or U2AF1^Q157R^ mutant expressing HSCs, consistent with U2AF1 being a haplo-essential gene,^18^ and reinforcing that the U2af1^Q157R^ allele impairs U2AF1 function despite the less severe phenotype compared to U2af1^S34F^.
U2AF1 S34F and U2AF1Q157R induce distinct gene expression changes in mouse myeloid progenitor cells.
To characterize the effects of mutant U2AF1 on gene expression in vivo, we performed RNA-seq of total RNA (rRNA-depleted) from BM myeloid progenitor (KL) cells from U2af1^Q157R/+^, U2af1^S34F/+^, or U2af1^+/+^ control mice (all Mx1-Cre^+^) under native hematopoiesis conditions (as in Fig. 1D). KL cells were isolated by FACS at 4 weeks after completion of pIpC injections and the variant allele frequencies of the S34F and Q157R mutations were near 50% (Supplementary Fig. 5A). Unsupervised principal component analysis of gene expression values (N = 19312 genes) segregated U2af1^Q157R/+^, U2af1^S34F/+^, and U2af1^+/+^ KL cells (Fig. 5A and Supplementary Table 3). Reanalysis of U2af1^S34F/+^ Native KL RNA-seq data published by Fei et al.^17^ demonstrated a strong concordance in gene expression changes with our U2af1^S34F/+^ KL data (Supplementary Fig. 5B,C). In our dataset, we identified 185 differentially expressed genes (DEGs; FDR<0.05 and ∣log_2_FC∣>1) in U2af1^S34F/+^ compared to U2af1^+/+^ control mice (Fig. 5B) and 77 DEGs in U2af1^Q157R/+^ KL cells (Fig. 5C). There were only 12 DEGs shared between U2af1^S34F/+^ and U2af1^Q157R/+^ KL cells (4.8%; Fig. 5D) with no overlap in gene ontology (GO) analysis (Supplementary Fig. 5D,E and Supplementary Table 4). Gene set enrichment analysis (GSEA) revealed significant positive enrichment of the p53 pathway in U2af1^S34F/+^ KL cells and negative enrichment of immune response related Hallmark pathways in both U2af1^S34F/+^ and U2af1^Q157R/+^ KL cells compared to U2af1^+/+^ KL cells (Fig. 5E).
U2AF1 S34F and U2AF1Q157R induce distinct alternative pre-mRNA splicing changes in myeloid progenitor cells.
Using the same bulk RNA-seq data, we next characterized the effects of mutant U2AF1 on alternative mRNA splicing in vivo. We employed replicate multivariate analysis of transcript splicing (rMATS)^20^ to assess differential alternative pre-mRNA splicing of five different types of annotated splicing events (alternative 3’ or 5’ splice sites [A3SS, A5SS], mutually exclusive exons [MXE], retained introns [RI], and skipped exons [SE]) in U2af1^Q157R/+^, U2af1^S34F/+^, and U2af1^+/+^ KL cells. Unsupervised principal component analysis of inclusion ratios (referred to as ‘percent spliced-in’ or ‘PSI’) for all annotated alternative splicing events (N = 11580) revealed that global alternative pre-mRNA splicing is distinct between U2af1^Q157R/+^, U2af1^S34F/+^, and U2af1^+/+^ KL cells (Fig. 6A and Supplementary Tables 5–7).
We then applied rMATS to identify 1048 and 580 differentially spliced events (DSEs; FDR < 0.05 and ∣ΔPSI∣>0.05 vs U2af1^+/+^) in U2af1^S34F/+^ and U2af1^Q157R/+^ KL cells, respectively (Fig. 6B and Supplementary Table 8). We also applied our rMATS analysis pipeline to the U2af1^S34F/+^ Native KL RNA-seq dataset published by Fei et al.^17^ (Supplementary Fig. 6A,B) and observed a strong concordance (i.e., unidirectional ΔPSI values) between DSEs shared between the two U2af1^S34F/+^ KL datasets (Supplementary Fig. 6C). Thus, rMATS analysis of independent RNA-seq data demonstrates that the U2af1^S34F/+^ mouse model produces robust and reproducible gene expression and alternative pre-mRNA splicing changes in hematopoietic cells in vivo (Supplementary Figs. 5B,6C). In line with previous studies of U2AF1 mutant cell lines and patient HSPC, SE events represented the majority of DSEs identified in U2AF1 mutant mouse KL cells (Fig. 6B and Supplementary Fig. 6A).^17,21,22^ U2af1^S34F/+^ SE DSEs also favored exon exclusion (‘skipping’) over exon inclusion.^23^ Of note, U2af1^Q157R/+^ DSEs were more equally distributed between RI and SE events (Fig. 6B). The overlap of DSE shared between U2af1^S34F/+^ and U2af1^Q157R/+^ KL cells was low (125 events or 8.3%; Fig. 6C). Conversion of DSE to differentially spliced genes (DSG) revealed 196 genes (17.5%) aberrantly spliced in common between the two mutants (Fig. 6D). GO analysis revealed that DSGs from U2af1^S34F/+^ KL cells were most significantly enriched in mRNA binding and metabolism gene sets, as well as histone post-translational modification and stress granule^23^ related gene sets (Supplementary Fig. 6D and Supplementary Table 9). DSGs from U2af1^Q157R/+^ KL cells were enriched in mRNA gene sets to a weaker extent than U2af1^S34F/+^ (Supplementary Fig. 6D and Supplementary Table 9).
Analysis of consensus 3’ splice site (3’SS) sequences from differentially spliced SE events in U2af1^S34F/+^ and U2af1^Q157R/+^ KL cells confirmed previous dependencies identified in U2AF1 mutant cell lines and patient HSPC. Specifically, exon inclusion favored a C and exon exclusion favored a T at the – 3 position of the 3’SS in U2af1^S34F/+^ cells (Fig. 6E, middle). In contrast, exon inclusion favored a G and exon exclusion favored an A at the + 1 position of the 3’SS in U2af1^Q157R/+^ cells (Fig. 6E, right). Overall, these findings highlight that the U2AF1^S34F^ and U2AF1^Q157R^ mutants induce significant but distinct changes to alternative mRNA splicing in vivo.
U2af1 S34F/+ and U2af1Q757R/+ mouse models recapitulate alternative pre-mRNA splicing changes found in MDS and AML patients.
To assess how well alternative splicing changes in mouse KL cells recapitulate changes seen in MDS and AML patient hematopoietic cells, we performed a meta-analysis using publicly available RNA-seq data from three published studies.^9,11,24^ Each study included 2–10 U2AF1^S34F^ and only 1–2 U2AF1^Q157R^ patients. Therefore, U2AF1^R156H^ and U2AF1^Q157(P/R)^ patient samples were grouped together (N=4–5 U2AF1^R156H/Q157(P/R)^ patients per study; Fig. 7A) consistent with previous studies demonstrating similar 3’SS sequence dependencies.^21,22^ In each study, samples from MDS/AML patients who did not have identifiable mutations in SF3B1 or SRSF2 were used as a comparator (Splicing Factor [SF]^WT^). To allow for a more rigorous analysis of differential splicing, we reanalyzed the FASTQ files for each study using the same analysis workflows and applied the same significance thresholds (FDR < 0.05 and ∣ΔPSI∣>0.05 vs SF^WT^) as used for the analysis of mouse KL cells. Using this approach, we credentialed each of the three datasets (referred to as Madan,^11^ Pellagatti,^9^ and Beat AML^24^) (Fig. 7A and Supplementary Fig. 7A-I and Supplementary Tables 10–15). Specifically, SE events were the most frequent DSE type identified in each study for S34F and R156/Q157 (Supplementary Fig. 7A-C) and these events favored the characteristic consensus 3’SS sequence dependencies identified previously (Supplementary Fig. 7G-I).^9,17,21,22,25,26^ To increase rigor of our meta-analysis we prioritized only the DSEs that were shared between at least two of the three MDS/AML datasets for either U2AF1^S34F^ or U2AF1^R156/Q157^ (Fig. 7B and Supplementary Table 16). The distribution of these DSEs was similar to each individual dataset with SE events still representing the majority event type in U2AF1 mutant MDS/AML cells (Fig. 7C). As in the mice, the overlap of DSE shared between U2AF1^S34F^ and U2AF1^R156/Q157^ MDS/AML cells was low (144 of 1978 events or 7.3%; Fig. 7D). Conversion of DSE to DSG revealed a total of 284 of 1305 genes (21.8%) aberrantly spliced in common between the two mutants (Fig. 7E).
The overlap of DSG identified in human and mouse cells revealed that approximately 20% of aberrantly spliced genes in KL (mouse) cells were also mis-spliced in MDS/AML (human) cells for both U2AF1^S34F^ (17.6% shared) and U2AF1^Q157R^ (19.7% shared) mutants (Fig. 7F,G). GO analysis revealed that shared S34F DSGs were most significantly enriched in mRNA binding and metabolism gene sets as well as stress granule^23^ and mRNA translation related gene sets (Fig. 7H and Supplementary Table 17). Shared Q157R DSGs were less significantly enriched in mRNA gene sets than S34F. Histone binding and DNA damage response gene sets were among some of the significantly enriched gene sets for Q157R DSGs (Fig. 7H and Supplementary Table 17).
We validated several of these putatively shared aberrant splicing changes identified by rMATS analysis by performing RT-PCR followed by gel electrophoresis of RNA isolated from additional mouse KL cell samples (N = 4 per genotype) and MDS patient samples (N = 4–9 per genotype). Consistent with previous observations,^17,25-28^ we confirmed aberrant splicing of functionally relevant transcripts (H2AFY and GNAS) in U2AF1^S34F^ mutant mouse KL and MDS cells (Fig. 7I,J). We also demonstrate that aberrantly spliced transcripts (MPHOSPH9, SETD5, ATP6V0A7, and CLIP7) in U2AF1^Q157R^ mutant MDS patient cells are similarly mis-spliced in KL cells from U2af1^Q157R/+^ mice (Fig. 7I,K-L). Aberrant splicing of CLIP1 is one example of an SE event that is differentially spliced in opposite directions by U2AF1^S34F^ (increased exon inclusion) and U2AF1^Q157R^ (increased exon skipping/exclusion) in mouse and human cells (Fig. 7L), further highlighting the distinct splicing differences induced by these two U2AF1 mutants.
U2AF1 R156/Q157 mutations are enriched in patients with CMML and MPN compared to U2AF1S34F mutations.
Given the differences in gene expression, splicing, and hematopoietic phenotypes induced by U2af1^S34F/+^ and U2af1^Q157R/+^ mutations in mice, we asked if the two hotspot mutations were differentially enriched in various myeloid neoplasms. We identified 487 patients with a diagnosis of AML, sAML (from MDS), MDS, CMML, or MPN who had a U2AF1 mutation based on available sequencing data and calculated the proportion of patients with U2AF1^R156/Q157^ or U2AF1^S34^ mutations (see Supplementary Methods). We observed that U2AF1^R156/Q157^ mutations were more common in CMML and MPN patients, U2AF1^S34^ mutations more common in sAML and AML patients, and a similar proportion of both mutations occurred in MDS (Fig. 8A and Supplementary Table 18). The co-occurrence of U2AF1 and signaling gene mutations also differed across myeloid neoplasms, with NRAS and FLT3 mutations being more common with S34 mutations and CBL, PTPN11 and CSF3R mutations more common with R156/Q157 mutations. Similar to previous reports by our group and others, we also observed preferential co-occurrence of other gene mutations with U2AF1^R156/Q157^ (e.g., ASXL1) or U2AF1^S34F^ (e.g., BCOR) mutations in MDS patients (Fig. 8B and Supplementary Tables 19–20).^15,16^
DISCUSSION
In this study, we characterized the in vivo consequences of expressing two myeloid neoplasm-associated hotspot mutations in U2AF1 that code for S34F and Q157R substitutions. Our results indicate that the two mutations induce distinct hematopoietic phenotypes in mice, suggesting that the U2af1^S34F^ and U2af1^Q157R^ mutations should not be conflated as they may impact disease pathogenesis differently in patients. Mice expressing U2af1^S34F^ have a more severe reduction in their PB and BM cell counts, and reduced HSPCs repopulating ability, compared to mice expressing U2af1^Q157R^. The expression and splicing of the majority of target genes are unique between the mutations, in both mouse and human samples, potentially driving the phenotypic differences induced by the two mutations. The two mutations co-occur with different gene mutations and are not equally represented in various myeloid neoplasms, suggesting that multiple mechanisms are likely to drive the pathogenesis of U2AF1 mutant myeloid diseases.
Our results add to the growing body of literature highlighting the paradigm that different hotspot mutations in a specific cancer gene can lead to distinct functional consequences and should, therefore, not necessarily be conflated. In one of the more well studied examples, different KRAS hotspot mutations (e.g., G12, G13, Q61) lead to varying levels of KRAS activation through modulation of distinct biochemical properties of KRAS.^29^ In turn, different KRAS hotspot mutations confer different prognostic value in various cancers (e.g., colorectal cancer) and are predictive of response to chemotherapy and/or targeted therapies.^29^ Similarly, the prognostic significance of distinct SF3B1 mutations can be different in MDS, including their impact on overall survival.^30,31^ With respect to U2AF1, a prognostic scoring model for MF (MIPSS70 + v2.0) now incorporates the negative impact of Q157 (but not S34) codon mutations.^32^ U2AF1 S34 and Q157 codon-specific clinical characteristics have also been reported in MDS.^33,34^ Further studies are needed to fully understand the functional impact of each U2AF1 hotspot mutation in patients and determine whether these differences confer consistent prognostic or therapeutic value across the spectrum of myeloid malignancies.
Enrichment of U2AF1^S34^ vs U2AF1^Q157^ mutations in different myeloid diseases suggest that mutations may contribute to the disease phenotype by differences in the target genes that they dysregulate and/or cooperating gene mutations. Identifying and validating the key target genes that are dysregulated and confer mutation-specific cellular phenotypes will require future in vivo functional studies. Additionally, based on differences in hotspot mutations in other cancers, the cellular ‘soil’ that a S34 or Q157 mutation occurs in likely also matters. In U2AF1-mutated solid tumors, particularly lung adenocarcinomas and endometrial cancers, S34 codon mutations are highly enriched compared to Q157 codon mutations.^35^ This observation is not specific to U2AF1, as SF3B1 R625 codon mutations are enriched in uveal and cutaneous melanomas, whereas K700 mutations are more common in breast cancer and chronic lymphoid leukemia specimens.^35^ The subtle phenotype in the U2af1^Q157R/+^ mouse, including lack of severe cytopenias and possibly a slight increase in platelets, could make Q157R cells more permissive to transformation with a MPN-associated cooperating mutation (e.g., CSF3R) resulting in higher blood counts, something that will require future studies. In contrast, S34F induces cytopenias in mice and may contribute to cytopenias seen in MDS. In addition, S34 and Q157 do not cooperate with the same mutations in MDS (e.g., BCOR with S34 > Q157 and ASXL1 with Q157 > S34),^36^ and this could impact mutation-associated phenotypes in patients.^15,37^ Finally, U2AF1 hotspot mutation phenotypes could be influenced by the order of cooperating gene mutation acquisition (i.e., U2AF1 mutation occurring before or after a cooperating gene mutation) or the presence of hematopoietic stressors, requiring future experiments.
Collectively, our results support that U2AF1^S34F^ and U2AF1^Q157R^ mutations induce distinct hematopoietic, gene expression, and RNA splicing phenotypes in vivo. Larger population studies will be needed to determine if these phenotypic changes translate into clinico-pathologic differences in patients warranting separate classification.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Damm F, Kosmider O, Gelsi-Boyer V, Mutations affecting m RNA splicing define distinct clinical phenotypes and correlate with patient outcome in myelodysplastic syndromes. Blood. 2012;119(14):3211–3218.22343920 10.1182/blood-2011-12-400994 · doi ↗ · pubmed ↗
- 2Haferlach T, Nagata Y, Grossmann V, Landscape of genetic lesions in 944 patients with myelodysplastic syndromes. Leukemia. 2014;28(2):241–247.24220272 10.1038/leu.2013.336PMC 3918868 · doi ↗ · pubmed ↗
- 3Papaemmanuil E, Gerstung M, Malcovati L, Clinical and biological implications of driver mutations in myelodysplastic syndromes. Blood. 2013;122(22):3616–3627; quiz 3699.24030381 10.1182/blood-2013-08-518886 PMC 3837510 · doi ↗ · pubmed ↗
- 4Barraco D, Elala YC, Lasho TL, Molecular correlates of anemia in primary myelofibrosis: a significant and independent association with U 2AF 1 mutations. Blood Cancer J. 2016;6(4):e 415.27058230 10.1038/bcj.2016.22PMC 4855254 · doi ↗ · pubmed ↗
- 5Kandarpa M, Robinson D, Wu Y-M, Broad next generation integrated sequencing of myelofibrosis identifies disease-specific and age-related genomic alterations. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2024;10.1158/1078-0432.CCR-23-0372 PMC 1106160238386293 · doi ↗ · pubmed ↗
- 6Palomo L, Meggendorfer M, Hutter S, Molecular landscape and clonal architecture of adult myelodysplastic/myeloproliferative neoplasms. Blood. 2020;136(16):1851–1862.32573691 10.1182/blood.2019004229 PMC 7645608 · doi ↗ · pubmed ↗
- 7Patel BJ, Przychodzen B, Thota S, Genomic determinants of chronic myelomonocytic leukemia. Leukemia. 2017;31(12):2815–2823.28555081 10.1038/leu.2017.164PMC 8370480 · doi ↗ · pubmed ↗
- 8Lindsley RC, Mar BG, Mazzola E, Acute myeloid leukemia ontogeny is defined by distinct somatic mutations. Blood. 2015;125(9):1367–1376.25550361 10.1182/blood-2014-11-610543 PMC 4342352 · doi ↗ · pubmed ↗