UNC13A Polymorphism Influences Survival in Patients with Frontotemporal Dementia
Lianne M. Reus, Sean W. Willemse, Sterre C.M. de Boer, Julie De Houwer, Willem L. Hartog, Merel O. Mol, Jeroen G. J. van Rooij, Niccolo Tesi, Laura Donker Kaat, Marc Hulsman, Everard G. B. Vijverberg, Henne Holstege, Wouter van Rheenen, Jan H. Veldink, Leonard H. van den Berg

TL;DR
A genetic variation in UNC13A is linked to shorter survival in patients with frontotemporal dementia.
Contribution
This study identifies a specific UNC13A polymorphism as a survival predictor in frontotemporal dementia patients.
Findings
UNC13A rs12608932-CC homozygosity is associated with shorter survival in FTD patients.
The hazard ratio for survival was 1.28 in UNC13A CC homozygotes compared to other genotypes.
The finding has implications for patient counseling and clinical trial design in FTD.
Abstract
UNC13A (rs12608932‐CC) is associated with both amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD), and shortens survival in ALS. We aim to describe the association for UNC13A and survival in FTD. We included 626 patients with FTD from Dutch memory clinics, including a subcohort of 150 patients with TDP‐43 pathology. Survival analyses were performed using Cox proportional hazard models in a recessive manner. Homozygosity for rs12608932‐C in UNC13A was associated with a shorter survival compared with other genotypes (hazard ratio [HR] = 1.28, 95% confidence interval [CI] = 1.02–1.60, p = 0.033), which has implications for patient counselling and trial design. ANN NEUROL 2025;97:1062–1066
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
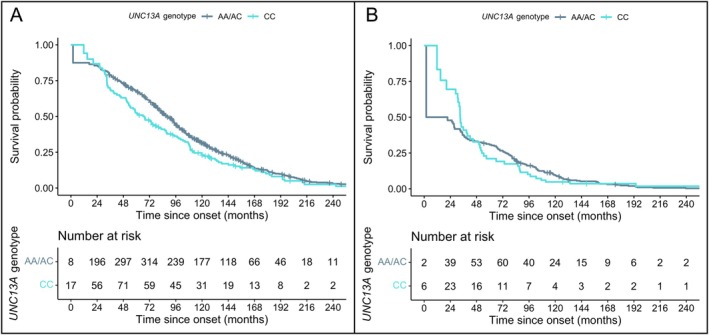 Figure 1
Figure 1| Total Cohort |
| TDP‐43 Cohort |
| |||||
|---|---|---|---|---|---|---|---|---|
| All (n = 626) | AA/AC (n = 512) | CC (n = 114) | All (n = 150) | AA/AC (n = 111) | CC (n = 39) | |||
| Male, n (%) | 356 (57) | 303 (59) | 53 (46) |
| 78 (52) | 62 (56) | 16 (41) | 0.16 |
| Age at study entry, yr, mean (SD) | 63.2 (8.2) | 63.3 (8.1) | 62.4 (8.7) | 0.28 | 62 (7.6) | 62.1 (7.9) | 61.6 (7.0) | 0.71 |
| Age at onset, yr, mean (SD) | 59.6 (8.4) | 59.6 (8.3) | 59.4 (8.7) | 0.81 | 58.4 (7.5) | 58.2 (7.8) | 59.1 (7.0) | 0.51 |
| Mendelian mutation, n (%) | 87 (14) | 70 (14) | 17 (15) | 0.84 | 87 (58) | 70 (63) | 17 (44) | 0.053 |
| Died, n (%) | 488 (78) | 394 (77) | 94 (82) | 0.25 | 136 (91) | 99 (89) | 37 (95) | 0.47 |
| Survival since onset, mo, median [95% CI] | 102 [72.0–141.3] | 105.5 [76.6–145.4] | 87.8 [54.5–126.2] |
| 85.2 [52.5–119.3] | 90 [68.4–123.0] | 52.4 [33.9–88.7] |
|
| N | HR [95% CI] |
| |
|---|---|---|---|
| Total cohort | 626 | 1.28 [1.02–1.60] |
|
| Total excluding Mendelian mutations | 539 | 1.29 [1.01–1.65] |
|
|
| 59 | 0.99 [0.41–2.37] | 0.97 |
| Clinical subtype | |||
| bvFTD | 350 | 1.38 [1.03–1.84] |
|
| svPPA | 104 | 1.11 [0.5–2.49] | 0.79 |
| nfvPPA | 54 | 0.82 [0.4–1.69] | 0.60 |
| FTD‐ALS | 36 | 1.43 [0.67–3.03] | 0.35 |
| Pathological subtype | |||
| TDP‐43 | 150 | 1.52 [1.03–2.26] |
|
| Tau | 32 | 1.58 [0.42–6.0] | 0.50 |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsAmyotrophic Lateral Sclerosis Research · Prion Diseases and Protein Misfolding · Alzheimer's disease research and treatments
Frontotemporal dementia (FTD) is the second most common young‐onset dementia, characterized by behavioral changes and aphasia as main symptoms.1 FTD is part of a disease continuum with amyotrophic lateral sclerosis (ALS), a neurodegenerative disease characterized by motor neuron loss in the spinal cord and brain leading to spasticity, progressive weakness, and respiratory failure. Approximately 5 to 15% of patients with FTD develop motor neuron involvement, whereas 5 to 10% of patients with ALS meet the diagnostic criteria for FTD and nearly half develop cognitive or behavioral deficits in the frontotemporal spectrum.2, 3
The discovery of TDP‐43 mislocalization and aggregation as a pathological hallmark for both FTD and ALS has explained the relation between these diseases. In FTD, TDP‐43 pathology is present in around 50% of cases, with the other half comprising of Tau pathology or, more rarely, fused‐in sarcoma (FUS).1 In ALS, approximately 97% of ALS cases are characterized by TDP‐43 pathology, with only a small percentage (~3%) showing FUS or superoxide dismutase 1 (SOD1) pathology.2
Besides shared pathological and clinical characteristics, there is a common genetic basis. Familial forms of ALS and FTD can be caused by rare pathogenic variants in a number of genes, including C9orf72, TBK1, and TARDBP. Genome‐wide association studies have also identified common genetic variants within an intron of UNC13A as a shared risk factor. The most significant association in both ALS and FTD is a single nucleotide polymorphism (SNP) at rs12608932. Subsequent studies in ALS have shown that homozygosity for the C‐allele at rs12608932 in UNC13A modifies the clinical phenotype, implicating shorter survival and a higher frequency of bulbar‐onset, cognitive impairment and FTD.4, 5, 6
Recently, the pathophysiological mechanism driving this effect has been elucidated. The relevant intron within UNC13A contains a cryptic exon, which is incorporated into mRNA under conditions of TDP‐43 pathology.7, 8 In turn, this leads to nonsense‐mediated decay of the mRNA in these cells, causing a loss of functional UNC13A and profound deficits in synaptic transmission.9
Considering UNC13A confers risk for both ALS and FTD and the effect on the ALS phenotype is TDP‐43 driven, we hypothesized that homozygosity for the C‐allele at rs12608932 in UNC13A negatively impacts survival in TDP‐43 associated FTD.
Methods
In total, 708 patients with FTD were selected from memory clinic cohorts in the Netherlands, including 290 patients from the Erasmus Medical Center (EMC) in Rotterdam and 418 from the Amsterdam Dementia Cohort (ADC). These cohorts have been described previously.10, 11 Both have been approved by a Medical Ethical Committee and all included patients or their carers have given informed consent for the use of their clinical and genetic data in scientific research. We collected the following clinical data: sex, age/time at study entry, age/time at onset, survival (defined as the time from first symptoms until censoring or death), date/time of censoring or date/time of death (taken from population registries), and clinical FTD subtype. Clinical subtyping included behavioral variant FTD (bvFTD), semantic variant primary progressive aphasia (svPPA), non‐fluent agrammatic primary progressive aphasia (nfvPPA) and FTD with motor neuron symptoms (FTD‐ALS). For both cohorts, patients were excluded if follow‐up data and/or genetic data (rs12608932 on UNC13A) were not available. Patients were genotyped with the Illumina Global Screening Array (GSA) and genotypes of rs12608932 were extracted. Quality control of the samples has been described in depth elsewhere.12
As we hypothesized that homozygosity for the C‐allele at rs12608932 in UNC13A (henceforth referred to as rs12608932‐CC) has an effect on survival in the presence of TDP43‐pathology, we excluded patients with postmortem pathology other than TDP‐43 and/or Mendelian mutation related to non‐TDP‐43 pathologies (n = 33). For 104 patients, postmortem pathology confirmation was available. The final total cohort included 626 patients. We further established a TDP‐43 subcohort. Patients were classified to the TDP‐43 subcohort if they carried a TDP‐43‐related Mendelian mutation, had TDP‐43 pathology postmortem, or had a clinical subtype indicative of TDP‐43 pathology (ie, FTD‐ALS), resulting in a TDP‐43 subcohort of 150 patients. The details for in‐ and exclusion of patients can be found in Supplementary Figure S1.
We performed survival analysis using Cox proportional hazard models with adjustment for left truncation, correcting for age at onset, sex, and study site. Median survival was calculated using a Kaplan–Meier estimate and presented with a 95% confidence interval (95% CI). We based groups on UNC13A genotype in a recessive manner (ie, rs12608932‐AA/AC vs CC). Additionally, we performed multiple sensitivity analyses: excluding Mendelian mutation carriers, stratifying according to clinical subtype, and including only patients with a known TDP‐43 burden. Due to pre‐planned subgroup analysis based on a singular hypothesis, we did not correct for multiple testing. Analysis were performed using R software version 4.3.1. The p values lower than 0.05 were considered significant.
Results
Demographic results of the total sample (N = 626, mean age = 63.2 ± 8.2 years, 43% female patients) are presented in Table 1. The rs12608932‐CC (n = 114) carriers did not differ from the other genotype group (n = 512 A/C and A/A) in terms of age at study entry, age at onset, and number of Mendelian mutation carriers. Comparisons on the 2 cohorts (ADC vs. EMC) are provided in Supplementary Table S1.
During 325.8 months of follow‐up, 488 patients (80%) died. The median survival for the total cohort was 102 months (95% CI = 72.0–141.3). For rs12608932‐CC, the median survival was 87.8 months (95% CI = 54.5–126.2) compared to 105.5 months (95% CI = 76.6–145.4) for other genotypes. The rs12608932CC was associated with a shorter survival when compared with other genotypes (hazard ratio [HR] = 1.28, 95% CI = 1.02–1.60, p = 0.033; see the Figure 1). The effect remained significant after exclusion of Mendelian mutation carriers (HR = 1.29, 95% CI = 1.01–1.65, p = 0.041; Table 2). In the TDP‐43 subcohort (n = 150), we also observed a shorter survival for rs12608932‐CC in comparison with other genotypes (median survival = 52.4 vs 90 months, HR = 1.52, 95% CI = 1.03–2.26, p = 0.036). After stratifying for clinical subtype, we only found a difference in survival for rs12608932‐CC carriers versus the other genotypes for bvFTD (HR = 1.38, 95% CI = 1.03–1.84, p = 0.03). Further results stratified for clinical and pathological subtype are shown in Table 2.
Survival of patients with FTD and patients with TDP‐43 pathology. Panel A shows overall survival of the total cohort, split for homozygosity for the C‐allele at rs12608932 in UNC13A in a recessive manner. Panel B shows overall survival of the TDP‐43 cohort, split for homozygosity for the C‐allele at rs12608932 in UNC13A in a recessive manner. The number at risk initially increases due to left truncation, where some individuals enter the study at later time points rather than at the onset. FTD = frontotemporal dementia. [Color figure can be viewed at www.annalsofneurology.org]
Discussion
In line with previous observations in ALS, we found a shorter survival of 17.7 months for patients with FTD homozygous for the C‐allele at rs12608932 in UNC13A in comparison to AA/AC‐genotypes. We were able to confirm this finding across multiple sensitivity analyses, showing the effect is independent of Mendelian mutations. Although the TDP‐43 cohort was smaller, the effect seemed stronger when compared with the total cohort with a survival difference of more than 35 months, which might support our assumption that the negative effect on survival is TDP‐43 dependent. Unfortunately, the number of patients with certain Tau‐associated FTD was limited and we are therefore unable to draw firm conclusions on whether the effect UNC13A has on survival is indeed absent under conditions of Tau pathology.
As 1 in 6 people is homozygous for rs12608932‐C, our findings yield implications for a large number of patients with FTD. Knowledge of UNC13A genotype holds prognostic value in clinical practice and it seems a factor of relevance, as survival in rs12608932‐CC is shorter compared to that previously described for genetic mutations.13 This seems especially true in the case of TDP‐43 pathology, with a difference in survival of more than 3 years. At present, discrimination between FTD with TDP‐43 or Tau pathology depends largely on autopsies. A recent finding that plasma derived extracellular vesicles can be used as biomarkers for TDP‐43 or Tau pathology would allow application of this knowledge while patients are alive.14 Furthermore, UNC13A genotype could be a factor to correct or stratify for in clinical trials in order to prevent skewed survival in a trial arm.
In addition, our findings further strengthen the commonality of FTD and ALS. The rs12608932‐CC in UNC13A is associated with a higher incidence of cognitive deficits and concomitant diagnosis of FTD when patients are diagnosed with ALS.6, 9 Additionally, patients with ALS with rs12608932‐CC have more cortical atrophy on magnetic resonance imaging (MRI) and more hypometabolism on positron emission tomography (PET) in the frontal and temporal lobes, when compared with other genotypes.6, 15 Further determination of the UNC13A phenotype in FTD is necessary in order to assess whether these findings are unique to rs12608932‐CC in ALS or are also present in such patients with FTD.
In summary, we find that homozygosity for the risk polymorphism at rs12608932 in UNC13A leads to shorter survival in patients with FTD. Future research elucidating the clinical phenotype of UNC13A in FTD and its similarities to ALS could warrant collective efforts for the development of UNC13A‐specific treatments.
Author Contributions
L.M.R., S.W.W., S.J.vdL., W.vR., J.H.V., L.H.vdB., M.A.vE., and Y.A.L.P. contributed to conception and design of the study; L.M.R., S.C.M.dB., J.D.H., W.L.H., M.O.M., J.G.J.vR., N.T., L.D.K., M.H., H.H., J.vC., H.S., S.J.vdL., and Y.A.L.P. contributed to the acquisition and analysis of data; L.M.R. and S.W.W. contributed to drafting the text or preparing the figures.
Potential Conflicts of Interest
Nothing to report.
Supporting information
Supplementary Figure S1: Flowchart with patient in‐ and exclusion for the total cohort and the TDP‐43 cohort. Panel A shows patient in‐ and exclusion for the total cohort. Panel B shows patient in‐ and exclusion for the TDP‐43 cohort.
Supplementary Table S1: Baseline statistics stratified for recruitment site.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Bang J , Spina S , Miller BL . Frontotemporal dementia. Lancet 2015;386:1672–1682.26595641 10.1016/S 0140-6736(15)00461-4PMC 5970949 · doi ↗ · pubmed ↗
- 2van Es MA , Hardiman O , Chio A , et al. Amyotrophic lateral sclerosis. Lancet 2017;390:2084–2098.28552366 10.1016/S 0140-6736(17)31287-4 · doi ↗ · pubmed ↗
- 3Burrell JR , Halliday GM , Kril JJ , et al. The frontotemporal dementia‐motor neuron disease continuum. Lancet 2016;388:919–931.26987909 10.1016/S 0140-6736(16)00737-6 · doi ↗ · pubmed ↗
- 4Diekstra FP , van Vught PWJ , van Rheenen W , et al. UNC 13A is a modifier of survival in amyotrophic lateral sclerosis. Neurobiol Aging 2012;33:630.e 3–630.e 8.10.1016/j.neurobiolaging.2011.10.02922118904 · doi ↗ · pubmed ↗
- 5van Rheenen W , van der Spek RAA , Bakker MK , et al. Common and rare variant association analyses in amyotrophic lateral sclerosis identify 15 risk loci with distinct genetic architectures and neuron‐specific biology. Nat Genet 2021;53:1636–1648.34873335 10.1038/s 41588-021-00973-1PMC 8648564 · doi ↗ · pubmed ↗
- 6Tan HHG , Westeneng HJ , van der Burgh HK , et al. The distinct traits of the UNC 13A polymorphism in amyotrophic lateral sclerosis. Ann Neurol 2020;88:796–806.32627229 10.1002/ana.25841 PMC 7540607 · doi ↗ · pubmed ↗
- 7Brown AL , Wilkins OG , Keuss MJ , et al. TDP‐43 loss and ALS‐risk SN Ps drive mis‐splicing and depletion of UNC 13A. Nature 2022;603:131–137.35197628 10.1038/s 41586-022-04436-3PMC 8891020 · doi ↗ · pubmed ↗
- 8Ma XR , Prudencio M , Koike Y , et al. TDP‐43 represses cryptic exon inclusion in the FTD–ALS gene UNC 13A. Nature 2022;603:124–130. 10.1038/s 41586-022-04424-7.35197626 PMC 8891019 · doi ↗ · pubmed ↗