Targeting Tumor Differentiation Grade-related Genes Prognostic Signature Including COL5A1 Based on Single-cell RNA-seq in Gastric Cancer
Jianming Wei, Xibo Gao, Zhufeng Li, Yangpu Jia, Chuan Li, Jian Liu

TL;DR
This study identifies a new set of genes, including COL5A1, that predict survival in gastric cancer patients based on tumor differentiation grade using single-cell RNA sequencing.
Contribution
A novel tumor differentiation grade-related gene signature, including COL5A1, is proposed as a prognostic biomarker in gastric cancer.
Findings
A tumor differentiation grade-related gene signature was established and validated for gastric cancer prognosis.
COL5A1 is significantly correlated with multiple immune cell types in gastric cancer.
The identified genes and risk score are significantly associated with patient survival.
Abstract
Background: Tumor differentiation grade was reported to be a prognostic factor in gastric cancer (GC). Here, we identify a novel tumor differentiation grade-related genes prognostic signature and provide new biomarkers using single-cell RNA sequencing (scRNA-seq) in GC. Methods: ScRNA-seq profiles of GC were analyzed by 'seurat' package. Tumor differentiation grade module was identified through a weighted gene co-expression network analysis (WGCNA). Hematoxylin and eosin (H&E) were performed to classify differentiation grade. The effects of tumor differentiation grade on prognosis were explored using the Kaplan-Meier. Tumor differentiation grade prognostic signature was constructed and validated in GC. Results: Using GEO database, the scRNA-seq cell differentiation, clusters, and marker genes were identified in GC. Functional enrichment analysis revealed that common differentially…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
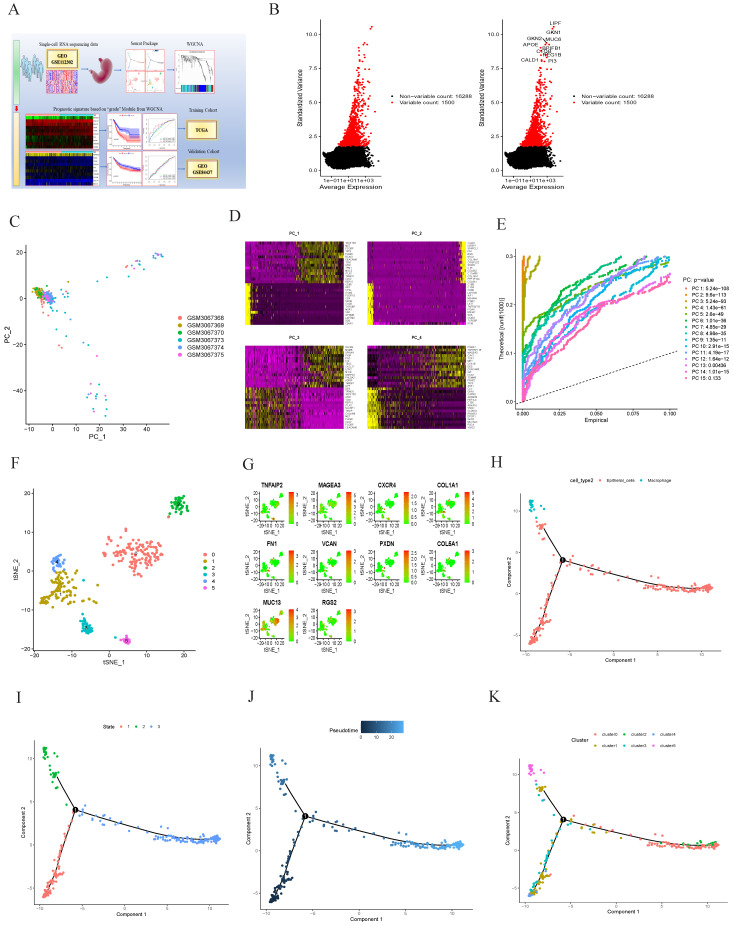 Figure 1
Figure 1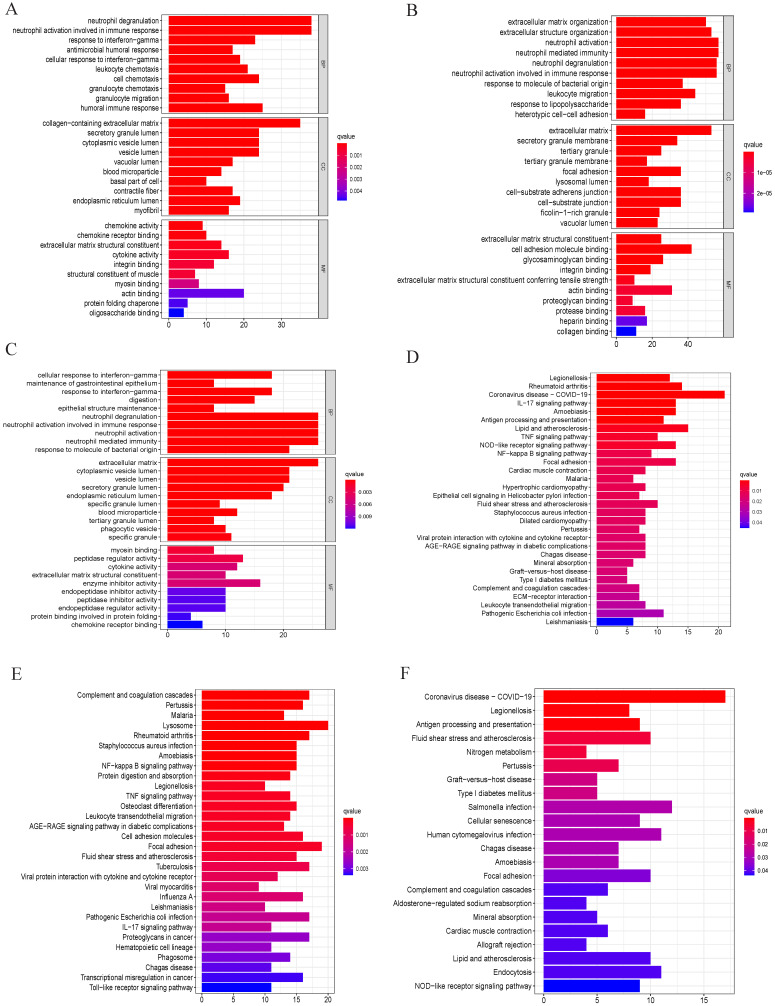 Figure 2
Figure 2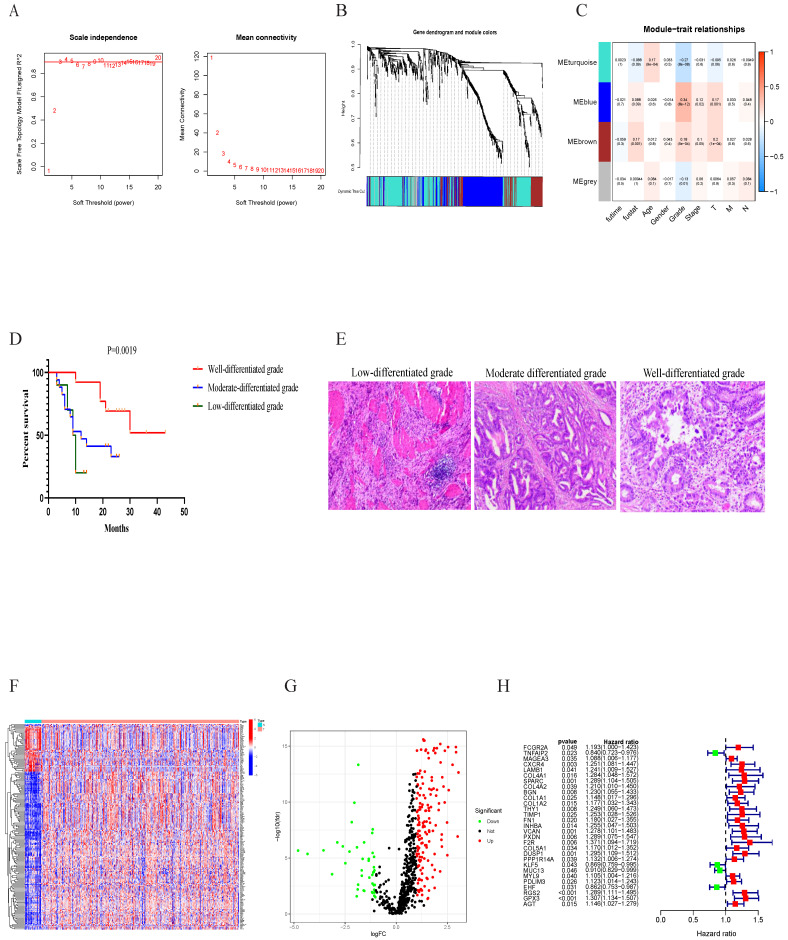 Figure 3
Figure 3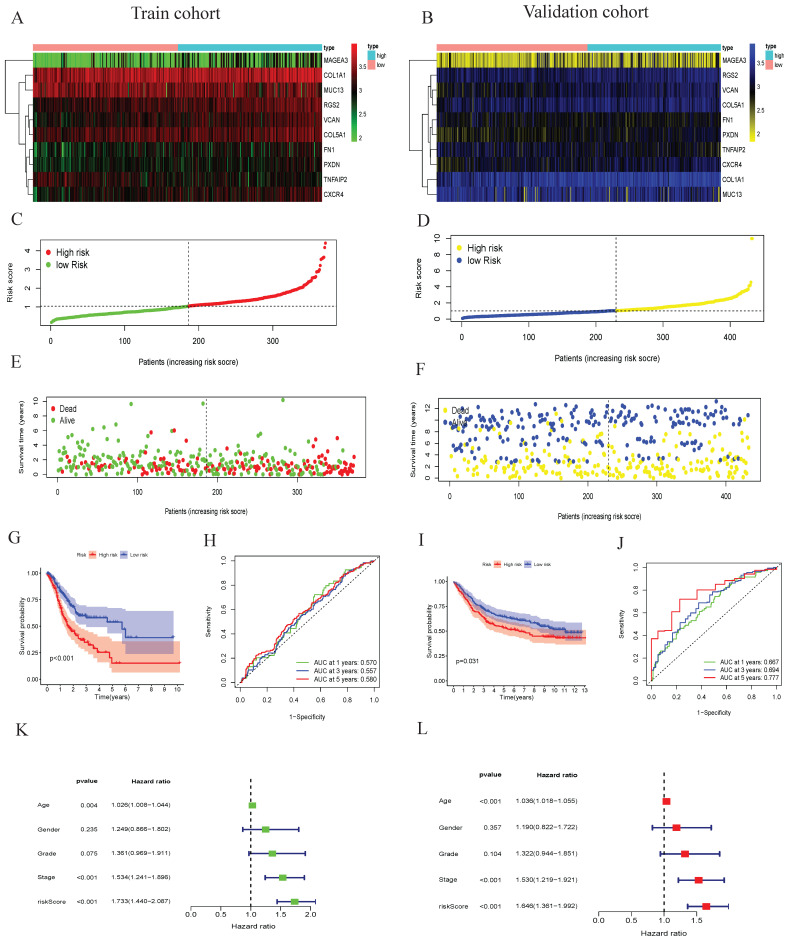 Figure 4
Figure 4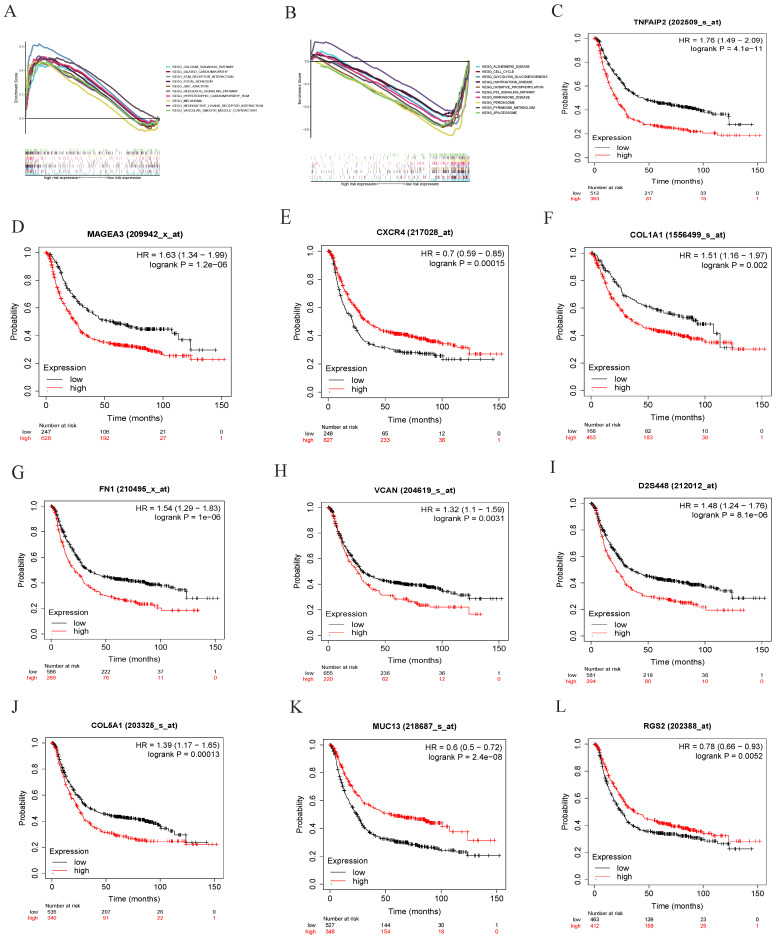 Figure 5
Figure 5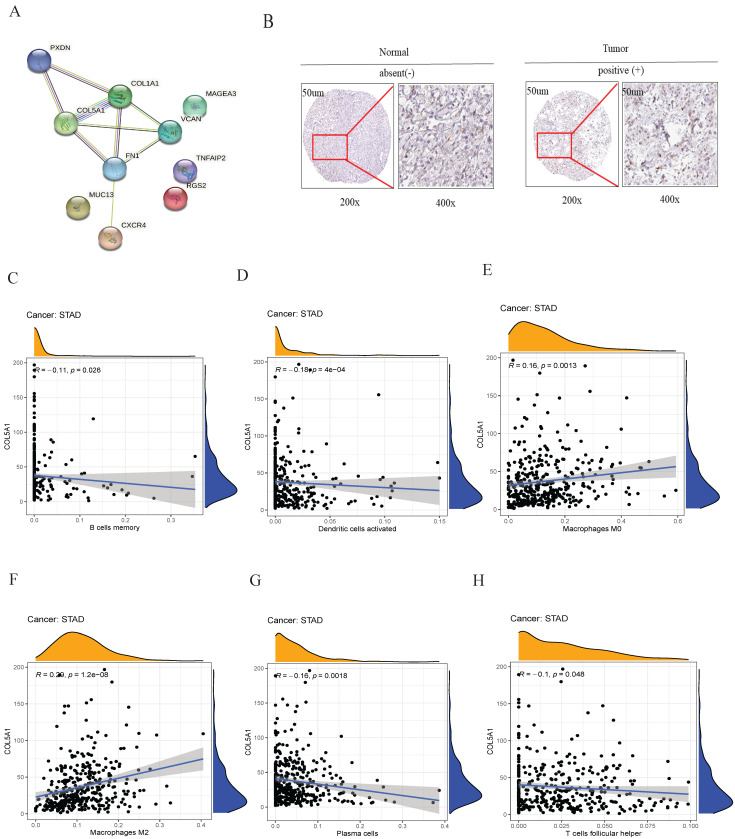 Figure 6
Figure 6Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsGastric Cancer Management and Outcomes · Cancer Genomics and Diagnostics · Esophageal Cancer Research and Treatment
Introduction
Gastric cancer (GC) is an aggressive tumor ranking fourth for mortality globally1, and its incidence rate is increasing over the recent decades. Although treatments with surgery and chemotherapy lead to a satisfactory survival benefit2, 3, the 5-year overall survival rate of GC is still low. Increasing biomarkers such as circular RNAs, microRNAs, and tumor microenvironment‑related genes have been identified to predict prognosis in GC4-6. Therefore, it is important to excavate the predictive biomarkers and find a novel strategy for GC.
With the increasing studies of cancer pathology, the tumor differentiation grade has been shown to be a prognostic factor in many cancers. Kristoffer derwinger et al. reported that the tumor differentiation grade was correlated significantly with the overall TNM stage and the risk of having lymph node metastasis7. A previous study had shown that apoptosis and cell proliferation correlated with tumor differentiation grade in patients with lung adenocarcinoma8. However, the prognostic risk model of tumor differentiation grade-related genes in GC was unexplored.
Single-cell RNA sequencing (scRNA-seq) is applied to explore the cellular components and gene expression at the single-cell level9, and can provide a new insight into tumor heterogeneity. Moreover, several studies have elucidated the association between TME and intra-tumoral heterogeneity using scRNA-seq10, 11. ScRNA-seq is also a powerful way for identifying anti-cancer drug response12, 13. However, the potential roles of the tumor differentiation grade-related genes and prognostic signature based on scRNA-seq have not been elucidated yet.
The present study develops a novel tumor differentiation grade-related genes prognostic signature which include ten genes (TNFAIP2, MAGEA3, CXCR4, COL1A1, FN1, VCAN, PXDN, COL5A1, MUC13 and RGS2). These genes are also highly connected in the protein-protein interaction (PPI) network. The hub gene COL5A1 is supposed to be significantly associated with tumor infiltrating immune cells and can be potential targets for prognosis in GC. Our data could support a deeper understanding of tumor differentiation grade-related genes in GC to provide appropriate therapy strategies.
Materials and methods
The study flowchart
The work flowchart is shown in Figure 1A, revealing the process of tumor differentiation grade-related genes prognostic signature construction based on scRNA-seq in GC.
Patient tissues and clinical data collection
Our study was approved by The Ethical Committee of Tianjin Medical University General Hospital (IRB2024-YX-595-01). Gastric tissues (n=40) previously collected by us, along with clinical data, were used. All histopathology were performed for evaluated tumor differentiation grade of GC in patients from January 2021 to August 2022.
Data processing
ScRNA-seq data from six primary gastric cancers and four normal gastric tissues were downloaded from GSE112302 dataset. Gene expression data was downloaded from GEO database including gastric cancer and normal tissues in GSE84437 for validation cohort. The original gene expression profiles and clinical data of GC in training cohort were obtained from the TCGA data portal. Firstly, the 'seurat' package was used to control data quality, and principal component analysis (PCA) was performed to conducted linear dimensionality reduction. Moreover, the t-Distributed Stochastic Neighbor Embedding (t-SNE) algorithm was applied to perform the cluster classification analysis and selected out the marker genes. Finally, the marker genes were annotated by the cluster and cell categories based on the 'SingleR' package (Version 0.99.13), and pseudotime analysis of cells was performed via the monocle package (Version 2.12.0).
Functional enrichment analysis
R package 'ggplot2', 'enrichplot', 'clusterProfiler' and 'GOplot' was applied to perform Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) GO and KEGG enrichment analysis. Functional annotation with a P-value <0.05 was considered statistically significant. Gene Set Enrichment Analysis (GSEA) is utilized for exploring signaling pathways in GC between the low and the high tumor differentiation grade risk.
The correlation of COL5A1 with tumor-infiltrating immune cells (TIICs)
CIBERSORT analysis tool is the deconvolution algorithm on basis of gene expression patterns for expressing tissue cell composition14. The 'CIBERSORT.R' package was performed to generate a proportion matrix of TIICs. The correlation of COL5A1 with TIICs was analyzed in this study.
Development and validation of tumor differentiation grade-related genes prognostic signature
WGCNA analysis was used to identify the gene association patterns between different samples and highly coordinated gene sets15. The significant modules related to clinical traits were screened out using WGCNA. DEGs based on tumor differentiation grade module from WGCNA analysis were used to establish a multivariable Cox regression model. The risk score formula was constructed according to the previous formula16.
Protein-protein interaction (PPI) and overall survival of tumor differentiation grade-related genes in GC
STRING database (https://cn.string-db.org/) was a functional protein association network, assembling all known and predicted proteins. The protein-protein interaction network of ten tumor differentiation grade-related genes was analyzed by STRING database. The Kaplan Meier plotter was applied to evaluate the correlation between the expression of all genes (mRNA, miRNA, protein) and survival in 30k+ samples from 21 tumor types17. The tool's primary purpose is the discovery and validation of survival biomarkers.
H&E staining
Histopathological examinations were performed by two experienced pathologists in our hospital. Three to four HE stained levels/sections were examined per specimen.
Statistical analysis
R software (version 4.0.0) was used to perform all statistical analyses in this study. Cox regression analysis, Kaplan-Meier curves with the log-rank test, receiver operating characteristic (ROC) curve the corresponding area under the ROC curve (AUC) were conducted by the 'glmnet', 'survival' and 'survivalROC' packages. Statistical significance was set at P < 0.05.
Results
ScRNA-seq profiling of the tumor landscape in primary GCs and normal gastric tissues
With scRNA-seq, 401 cell samples were acquired from 6 patient-derived GC tissues and 4 gastric normal tissues. Firstly, the study of quality control and data filtering was performed. Subsequently, the variance analysis revealed CALD1, PI3, REG1B, C1QB, APOE, BPIFB1, GKN2, MUC6, GKN1 and LIPF were the top ten significant DEGs in 1500 genes (Figure 1B). The PCA method was performed to screen out the significantly correlated genes in each component (Figure 1C-E). We also used the t-SNE algorithm successfully classified the samples into six clusters (Figure 1F). The expression of tumor differentiation grade-related genes prognostic signature in clusters (TNFAIP2, MAGEA3, CXCR4, COL1A1, FN1, VCAN, PXDN, COL5A1, MUC13 and RGS2) was shown in Fig.1G. Additionally, cell type was annotated for each cell sample, and all the cells in the six clusters were annotated as epithelial cells and macrophage (Figure 1H). Cell transition trajectory analyses showed that marker genes expression patterns changed in three ways, and we divided the cells into three branches: branch 1, branch 2, and branch 3. Branch 2 mainly represented macrophages. (Figure 1I-K).
Functional enrichment analysis of common DEGs from cell transition trajectory
To investigate the biological functional of common DEGs based on branch 1, branch 2 and branch 3, GO and KEGG were performed by R package 'ggplot2', 'enrichplot', 'clusterProfiler' and 'GOplot'. In this study, we found that neutrophil degranulation, neutrophil activation involved in immune response, neutrophil mediated immunity and neutrophil activation played an important role in biological processes (BP), cell components (CC), and molecular functions (MF) of GO terms (Figure 2A-C). In addition, we observed that common DEGs from cell transition trajectory were mainly enriched in coronavirus disease- COVID-19, legionellosis, rheumatoid arthritis, complement and coagulation cascades, pertussis, malaria, lysosome, antigen processing and presentation (Figure 2D-F). Above results showed that neutrophil could play an important role in GC.
Tumor differentiation grade-related genes identification
After data preprocessing, a total of 618 common DEGs from three branches were used to perform weighted gene co-expression network analysis (WGCNA). We also observed that 618 genes were also common in the expression matrix of GSE84437 and TCGA. The adjacency matrix and the topological overlap matrix were constructed (Figure 3A). Next, four modules were identified based on average hierarchical clustering and dynamic tree clipping (Figure 3B). Histological grading system revealed the extent of malignancy of GC. Jayanthi, V. et al. constructed grade-specific molecular interaction networks identified grade-specific biomarkers for breast cancer18. Then, we found that tumor differentiation grade was significantly associated with prognosis in GC. In this study, the blue module, brown module, grey module and turquoise module were highly related to tumor differentiation grade. Four modules were selected as a clinically important module for further analysis (Figure 3C). Kaplan-Meier analysis revealed that well differentiated grade had better prognosis in 40 GC patients (Figure 3D). H&E staining identified three group: well differentiated grade, moderate-differentiated grade, and low-differentiated grade (Figure 3E). Finally, a total of 190 differentially expressed genes were identified, as shown in Figure 3F, 3G.
We then integrated the clinical data from all patients of TCGA dataset, univariate analysis revealed that the expression of FCGR2A, TNFAIP2, MAGEA3, CXCR4, LAMB1, COL4A1, SPARC, COL4A2, BGN, COL1A1, COL1A2, THY1, TIMP1, FN1, INHBA, VCAN, PXDN, F2R, COL5A1, DUSP1, PPP1R14A, KLF5, MUC13, MYL9, PDLIM3, EHF, RGS2, GPX3, and AGT were distinctly associated with prognosis in GC patients (Figure 3H).
Risk model construction of tumor differentiation grade-related genes and external data validation
We then selected ten genes based on the coefficients derived from the multivariate analysis to construct risk model. The coefficients of the ten prognostic genes are shown in Table 1. The study formula for the risk score was as follows19: Risk score = ( -0.209406939 × expression value of TNFAIP2) + ( 0.126113147 × expression value of MAGEA3) + ( 0.171457823 × expression value of CXCR4) + ( 0.58121411 × expression value of COL1A1)+( -0.2694724× expression value of FN1 ) + ( 0.345428991 × expression value of VCAN)+( -0.800354177 × expression value of COL5A1)+( -0.098651917 × expression value of MUC13) + ( 0.345428991 × expression value of PXDN) + ( 0.202535842 × expression value of RGS2). To investigate genes expression profiles in high-risk and low-risk GC groups, gene heat map, the risk score distribution and follow-up time were shown in Figure 4A,4C,4E. The Kaplan-Meier plot revealed that patients in the high-risk group had a significantly poorer OS than those in the low-risk group in the train cohort (Figure 4G). The prognostic signature had a good accuracy to predict OS in the train cohort with 1-year, 3-year and 5-year AUC of 0.570, 0.557, 0.580 (Figure 4H).
The prognostic signature associated with grade module was validated using the same risk score formula and the same cut-off value. The gene expression pattern, risk distribution, survival status, the Kaplan-Meier plot and AUC were shown in Figure 4B, 4D, 4F, 4I, 4J. Multivariate Cox regression analyses showed that age, stage and the risk score were found to be independent risk factors for prognosis in patients with gastric cancer (Figure 4K-L). These analyses indicated that tumor differentiation grade-related genes and constructed risk prognostic models have good prognostic value.
GSEA enrichment and Kaplan-Meier Plotter analysis
In this study, GSEA software were applied to analyze the low-risk group and the high-risk group of tumor differentiation grade-related genes signature. We showed that grade-related genes in this signature are significantly enriched in the high-risk (Figure 5A) and low-risk groups (Figure 5B). The top five KEGG pathways in the high-risk group were neuroactive ligand receptor interaction, ECM receptor interaction, GAP junction, hypertrophic cardiomyopathy HCM, and focal adhesion. However, the top five KEGG pathways in the low-risk group were peroxisome, pyrimidine metabolism, glycolysis gluconeogenesis, alzheimers disease, and oxidative phosphorylation. Kaplan-Meier survival analysis suggested that the expression of TNFAIP2, MAGEA3, CXCR4, COL1A1, FN1, VCAN, PXDN, COL5A1, MUC13, and RGS2 had significantly worse prognosis (P < 0.05) (Figure 5C-L).
COL5A1 was significant differentially expressed and associated with immune cells
Among tumor differentiation grade-related genes in the prognostic signature, collagen type V alpha 1 (COL5A1) is a key molecular node (Figure 6A). Therefore, COL5A1, a risk factor of the risk model, was analyzed for subsequently experimental verification. We found that COL5A1 was highly expressed in the GC tissues compared to the adjacent normal tissues in the Human Protein Atalas webserver (Figure 6B). Subsequently, we observed that the expression of COL5A1 was significantly associated with B cells memory (Figure 6C), dendritic cells activated (Figure 6D), macrophages M0 (Figure 6E), macrophages M2 (Figure 6F), plasma cells (Figure 6G), T cells follicular helper (Figure 6H). These data showed that COL5A1 could play an important role in tumor microenvironment in GC.
Discussion
Morphological heterogeneity and genetic heterogeneity comprise tumor heterogeneity affecting diagnosis and therapy in cancer20. Gastric cancer is a highly heterogeneous malignant cancer with virous subtypes and clinical behaviors21, 22. Here, we explored tumor heterogeneity at the single cell level in GC using scRNA-seq.
Our result based on scRNA-seq analysis showed that tumors included virous cells, such as malignant cells, tumor infiltrating cells and stromal components. Cell transition trajectory analysis is widely used to explore different cell types at different stages of development and differentiation in several reports23, 24. We identified three branches in cell transition trajectory. In this study, common DEGs were analyzed in three branches. Our analysis revealed that these genes were enriched in Coronavirus disease 2019 (COVID-19). COVID-19 is defined as a respiratory tract infection caused by the severe acute respiratory syndrome (SARS) coronavirus (COV), also named SARS-CoV-225. COVID-19 was first found in Central China (Wuhan, the capital of Hubei province) at the end of December 201926. Many reports have explored the relationship of COVID-19 with cancer27-29. Hoang, T. et al reported that genetic susceptibility of ACE2 and TMPRSS2 in GC was associated with the susceptibility to COVID-19 infection30.
The tumor microenvironment (TME) consists of a heterogenous cellular component affecting cancer cell behavior. Sathe, A. et al showed that gastric cancer TME was significantly enriched for stromal cells, macrophages, dendritic cells (DC), and Tregs31. Our study also found that cells cluster 5 was main macrophages. We investigated that common DEGs in three branches were associated with neutrophil degranulation, neutrophil activation involved in immune response, neutrophil mediated immunity and neutrophil activation. Increasing studies revealed that neutrophil played a critical role in tumor microenvironment32-34.
In this study, we used ten tumor differentiation grade-related genes for the first time to establish the prognostic signature including TNFAIP2, MAGEA3, CXCR4, COL1A1, FN1, VCAN, PXDN, COL5A1, MUC13, and RGS2 based on single-cell RNA-seq. More importantly, age, stage and riskscore were independent prognostic factors. Using H&E, we evaluated tumor differentiation and found that patients in well differentiated grade group had better prognosis than other groups. To our knowledge, this is the first study that directly revealed that a prognostic risk model based on tumor differentiation grade-related genes could predict prognosis in GC.
Many recent studies showed that TNFAIP2, MAGEA3, CXCR4, COL1A1, FN1, VCAN, PXDN, COL5A1, MUC13, and RGS2 could predict overall survival as a single gene biomarker35-43. Here, we found that the expression level of TNFAIP2, MAGEA3, CXCR4, COL1A1, FN1, VCAN, PXDN, COL5A1, MUC13, and RGS2 is closely related to the prognosis of GC patients.
Several studies have used PPI network to study the interactome of protein and screen hub genes44. Our PPI network studies demonstrated that COL5A1 was a key molecular node. As the collagen family members, high level of COL5A1 is closely associated with the poor prognosis of multiple human tumors45, 46. Focused on the molecular role of COL5A1 in malignant cells, investigators have reported that NAT10 promoted GC metastasis via N4-acetylated COL5A147. We also observed that COL5A1 was differentially expressed in GC tissues. A previous study shows that COL5A1 was a cancer-associated fibroblast gene signature as a poor prognostic factor and potential therapeutic target in GC48. Given that the correlations of COL5A1 with immune infiltrating cells, our results revealed that COL5A1 was significantly correlated with B cell memory, dendritic cells-activated, macrophage M0, macrophage M2, plasma cells, and T cells follicular helper, suggesting its role in regulating TME. However, this study had no enough clinical samples to validate this prognostic signature using our experimental data.
Conclusions
Together, we screened tumor differentiation grade-related genes prognostic signature including NFAIP2, MAGEA3, CXCR4, COL1A1, FN1, VCAN, PXDN, COL5A1, MUC13 and RGS2, and provided evidence of GC heterogeneity based on single-cell RNA-seq. Importantly, COL5A1 may be as a novel therapeutic target and biomarkers for GC.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Sung H Ferlay J Siegel RL Laversanne M Soerjomataram I Jemal A Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries CA Cancer J Clin 202110.3322/caac.2166033538338 · doi ↗ · pubmed ↗
- 2Ahn SH Kang SH Lee Y Min SH Park YS Park DJ Long-term Survival Outcomes of Laparoscopic Gastrectomy for Advanced Gastric Cancer: Five-year Results of a Phase II Prospective Clinical Trial Journal of gastric cancer 201919102103094476310.5230/jgc.2019.19.e 6PMC 6441772 · doi ↗ · pubmed ↗
- 3Digklia A Wagner AD Advanced gastric cancer: Current treatment landscape and future perspectives World J Gastroenterol 2016222403142693712910.3748/wjg.v 22.i 8.2403 PMC 4768187 · doi ↗ · pubmed ↗
- 4Ghafouri-Fard S Vafaee R Shoorei H Taheri M Micro RN As in gastric cancer: Biomarkers and therapeutic targets Gene 20207571449373264030010.1016/j.gene.2020.144937 · doi ↗ · pubmed ↗
- 5Ouyang J Long Z Li G Circular RN As in Gastric Cancer: Potential Biomarkers and Therapeutic Targets Biomed Res Int 2020202027906793268545910.1155/2020/2790679 PMC 7345955 · doi ↗ · pubmed ↗
- 6Rojas A Araya P Gonzalez I Morales E Gastric Tumor Microenvironment Adv Exp Med Biol 2020122623353203067310.1007/978-3-030-36214-0_2 · doi ↗ · pubmed ↗
- 7Derwinger K Kodeda K Bexe-Lindskog E Taflin H Tumour differentiation grade is associated with TNM staging and the risk of node metastasis in colorectal cancer Acta Oncol 20104957622000150010.3109/02841860903334411 · doi ↗ · pubmed ↗
- 8Kalogeraki A Tzardi M Zoras O Giannikaki E Papadakis M Tamiolakis D Apoptosis and cell proliferation correlated with tumor grade in patients with lung adenocarcinoma In vivo 2010246677020952731 · pubmed ↗