Philadelphia chromosome-positive B-cell acute lymphoblastic leukemia with e13a3 fusion transcripts in a patient with pre-existing essential thrombocythemia: a case report and literature review
Ying Wu, Dan Cao

TL;DR
A rare case of a patient with essential thrombocythemia later developing Philadelphia chromosome-positive B-cell acute lymphoblastic leukemia is reported, with successful treatment and remission.
Contribution
First reported case of Ph-positive B-ALL with e13a3 fusion arising from pre-existing essential thrombocythemia.
Findings
Patient with essential thrombocythemia developed Ph-positive B-ALL with e13a3 fusion transcript.
Treatment with olverembatinib, vincristine, and prednisone achieved complete remission.
Consolidation therapy without chemotherapy maintained sustained molecular remission.
Abstract
Essential thrombocythemia (ET), a BCR-ABL1-negative myeloproliferative neoplasm (MPN), is characterized by persistent thrombocytosis and excessive megakaryocytic proliferation in the bone marrow. During the course of the disease, 4% of patients progress to acute leukemia, the majority of which have acute myeloid leukemia (AML), and are confirmed to be transformed from ET. Transformation to acute lymphoblastic leukemia (ALL) is exceedingly rare, with limited evidence clarifying its clonal relationship to antecedent ET. We report a case of a 66-year-old man with a history of ET, lacking mutations in Janus kinase 2 (JAK2), Calreticulin (CALR), or myeloproliferative leukemia virus oncogene (MPL), who subsequently developed Philadelphia chromosome (Ph)-positive B-cell ALL (B-ALL), harboring a rare e13a3 fusion transcript. Following four cycles of induction therapy with olverembatinib,…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
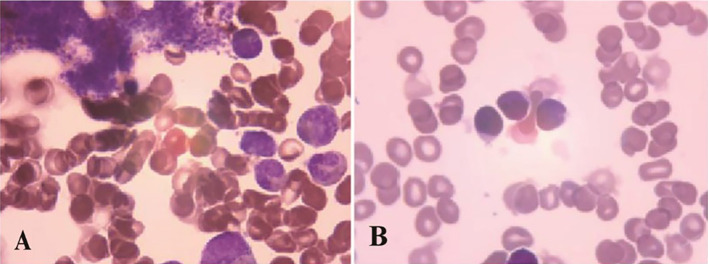 Figure 1
Figure 1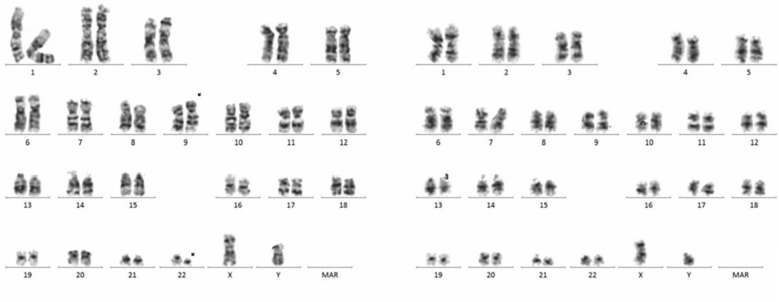 Figure 2
Figure 2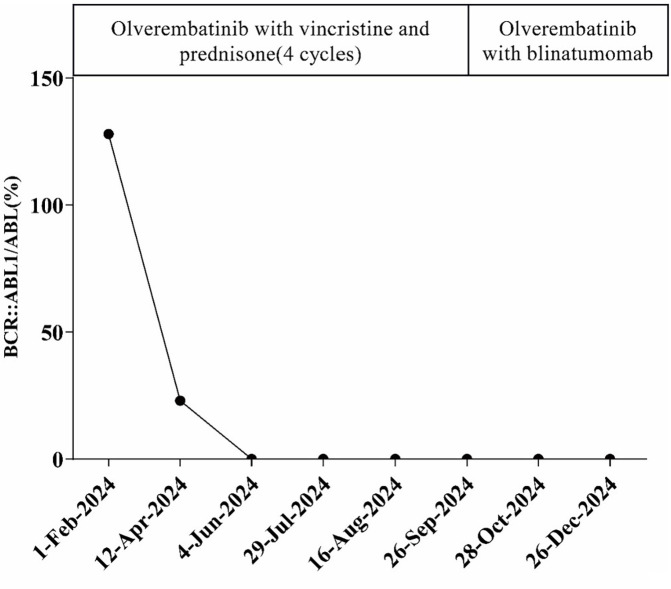 Figure 3
Figure 3| Our case | A M O'HEA et al | Berkahn et al | Nagai Y et al | Jiale Ma et al | Hilal T et al | Georgio Medawar et al | |
|---|---|---|---|---|---|---|---|
| age(year) | 66 | 61 | 87 | 67 | 71 | 69 | 49 |
| gender | male | female | female | female | female | female | male |
| pathogenic muation | negative | N/A | N/A | JAK2-V617F | N/A | CALR exon 9 | negative |
| treatment of ET | hydroxyurea and aspirin(2 years) | 5 mCi 32p | Hydroxyurea | Hydroxyurea (8years) and anti thrombotic agent (more than ten years) | hydroxyurea and antiplatelet therapy for 9 years | hydroxyurea, followed by anagrelide, and ruxolitinib for 1 month | Hydroxyurea |
| time of ET to ALL | 2 years | 16 years | 2 months | 16 years | 10 years | 26years | 10 years |
| ALL type | Ph+ ALL | B-ALL | T-ALL | Ph+ALL | B-ALL | B-ALL | T–ALL |
| Karyotype of leukemic transformation | 46,XY,t(9;22)(q34;q11.2)[19]/46,XY[1] | 44 XX with absent chromosomes 7, 21, and 14q+, 6p-. | 46, XX | Ph chromosome and monosomy 7 | 46,XX,t(9;22)(q34;q11)[8]/ 46,XX,[2] | 47,XX,t(1;19)(p13;p13.1), +der(1)t(1;19)[3]/46,XX[17]) | 46,XY,− 10,− 14,+2mar[6]/49~ 56, XY,+5,+15,+16,+19,+20,+20[cp2]/46, XY[12] |
| NGS |
| N/A | N/A | N/A |
| N/A |
|
| treatment of ALL | olverembatinib with VP(3 cycles) followed by olverembatinib and blinatumomab | supportive care | supportive care | dasatinib and prednisolone | flumatinib and prednisolone,followed by flumatinib maintenance | supportive care | Hyper-CVAD (4 cycles), followed by HSCT |
| outcome | alive with CMR | death after 5 months | rapid deterioration and death | relapsed with the T315I mutation nine months later | relapsed and died after 6 months | death after 3 months | remission at 4th month |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsChronic Myeloid Leukemia Treatments · Myeloproliferative Neoplasms: Diagnosis and Treatment · Acute Myeloid Leukemia Research
Introduction
Essential thrombocythemia (ET), a clonal disorder, arises from clonal expansion of hematopoietic stem cells, leading to sustained thrombocytosis and pathological megakaryocytic hyperplasia in the bone marrow (BM). Patients with ET face dual risks of thrombotic and hemorrhagic complications (1). While the median survival of ET is approximately 20 years, extending to 32.7 years for patients under 60 years, it remains reduced compared to age- and sex-matched healthy controls (2, 3). Compared to other myeloproliferative neoplasms (MPNs), leukemic transformation during the course of ET is rare, with a cumulative incidence of approximately 4%–5% over 20 years (4). However, it carries a dismal prognosis. Patients typically exhibit resistance to conventional chemotherapy, and allogeneic hematopoietic stem cell transplantation (allo-HSCT) remains the only curative option (5). Most patients with post-ET leukemia have acute myeloid leukemia (AML), with transformation to acute lymphoblastic leukemia (ALL) being exceptionally rare (5). The mechanism is not well understood and may include treatment-related factors, such as the use of hydroxyurea and alkylating agents, and high-risk gene mutations, including SRSF2, SF3B1, U2AF1, and TP53 (5, 6). Here, we report the first documented case of Philadelphia (Ph)-positive B-cell ALL (B-ALL) with e13a3 BCR::ABL1 fusion transcripts that followed triple-negative ET. Importantly, this patient achieved sustained complete molecular remission (CMR) through a chemotherapy-sparing regimen combining olverembatinib and blinatumomab, challenging the historical reliance on intensive chemotherapy or allo-HSCT in this high-risk population.
Case report
A 66-year-old male was incidentally found to have an elevated platelet count during a routine medical examination in 2022. The patient denied any clinical symptoms such as headaches, dizziness, chest pain, or paresthesia in the extremities. A complete blood count (CBC) showed a white blood cell count (WBC) of 6.2 × 10^9^/L, hemoglobin of 13.5 g/dL, and platelets of 674 × 10^9^/L. Physical examination was normal. The patient’s medical history was unremarkable for chronic illnesses, cardiovascular risk factors (hypertension, diabetes, or dyslipidemia), or familial predisposition to hematologic malignancies or inherited blood disorders. He denied tobacco use, alcohol consumption, or chronic exposure to medications or toxins. The BM aspiration showed an increased number of megakaryocytes with focal clustering (Figure 1A), while a biopsy showed 75% cellularity with grade 0 reticulin fibrosis. Megakaryocytes were large with hyperlobulated nuclei and abundant cytoplasm. No significant hyperplasia or left shift was observed in the granulocytic and erythroid series. Cytogenetic analysis revealed a normal diploid male karyotype. Molecular studies were negative for JAK2 V617F, CALR, and MPL mutations, as well as BCR::ABL1 fusion by reverse transcriptase-polymerase chain reaction (RT-PCR). ET was confirmed per WHO 2022 criteria: sustained thrombocytosis (≥450 × 10^9^/L); the exclusion of other myeloid neoplasms, including prefibrotic primary myelofibrosis (PMF), polycythemia vera, chronic myeloid leukemia, and myelodysplastic syndromes; megakaryocytic hyperplasia without dysplasia; and exclusion of reactive and secondary causes. The patient subsequently received treatment with hydroxyurea and aspirin, during which platelet counts fluctuated between 200–450 × 10^9^/L. In February 2024, the patient was admitted to the hospital due to diarrhea, fatigue, and fever for 5 days. A CBC at the local hospital showed a WBC of 67.7 × 10^9^/L, hemoglobin of 10.7 g/dL, and platelets of 46 × 10^9^/L. Peripheral blood smear revealed 9% abnormal cells. The patient was subsequently admitted to our hospital. A BM examination showed hypercellularity with increased numbers of blasts consistent with B-ALL (Figure 1B). Flow cytometry identified B-lymphoblasts accounting for approximately 91.489% of the leukocytes, expressing CD19, CD34, CD123, CD20, CD22, CD10, nTdT, cyCD79a, CD81, and CD9, but lacking CD117, CD7, CD13, CD33, CD38, CD56, CD5, sIgM, CD200, CDC66c, and cIgM. Karyotype analysis showed 46, XY, t(9;22)(q34; q11.2)[19]/46,XY[1] (Figure 2).Molecular studies confirmed the e13a3 BCR::ABL1 transcript. A 365-panel next-generation sequencing (NGS) revealed two mutations: SETD2 c.5126_5127insCC [variant allele frequency (VAF) 89.4%] and BTK c.1408A > G (VAF 1.5%). Thus, a diagnosis of Ph+ B-ALL was confirmed. At the time of the ET diagnosis (2022), the patient’s medical history was unremarkable for chronic illnesses or cardiovascular risk factors. However, during hospitalization for B-ALL progression in 2024, the patient developed acute chest pain. Comprehensive cardiac evaluation, including an electrocardiogram (ECG), cardiac biomarkers, echocardiography, and coronary computed tomography angiography (CTA), revealed ST-segment depression, troponin elevation, and microvascular dysfunction. A cardiology consultation confirmed the diagnosis of unstable angina. The patient was managed with dual antiplatelet therapy (aspirin and clopidogrel), atorvastatin, and isosorbide mononitrate. Given his age, unstable angina, and preference for minimal treatment disruption, we opted for a chemotherapy-sparing regimen combining olverembatinib (40 mg every other day) with vincristine and dexamethasone. One month later, a repeat bone marrow examination indicated morphological remission, and minimal residual disease (MRD) of 0.232% (1293/557378) by flow cytometry. Karyotype analysis showed 46,XY,t(9;22)(q34;q11.2)[12]/46,XY[8]. BCR::ABL1 transcript levels decreased to 22.9%. The patient declined allo-HSCT. Following three consolidation cycles with olverembatinib and VP (vincristine and prednisone), sustained CMR was achieved. The patient is currently receiving maintenance therapy with olverembatinib and blinatumomab (a bispecific anti-CD3/anti-CD19 T cell engager), and the disease remains in molecular remission. The treatment regimens and percentage of e13a3 fusion transcripts are illustrated in Figure 3. The patient will undergo monthly BCR::ABL1 PCR monitoring for 12 months, transitioning to quarterly assessments if the remission is sustained. A bone marrow evaluation (morphology, flow cytometry, and cytogenetics) will be carried out every 3 months for 2 years.
Bone marrow examination results: (A) 2022-1-11, morphological feature of ET (B) 2024-2-21, morphological feature of ALL.
Chromosome analysis: 46,XY,t(9;22)(q34;q11.2)[19]/46,XY[1].
Treatment regimen and the percentage of BR::ABL1 transcripts during the course of the disease.
Discussion
Leukemic transformation in ET is uncommon, with an estimated 10-year cumulative incidence of <1% (7). Recognized risk factors include JAK2 mutations, chromosomal abnormalities, and extreme thrombocytosis (platelet count ≥1000 × 10^9^/L) (8, 9). Most patients transform to AML and transformation to ALL is rare, with only case reports available. O’Hea et al. reported a case of B-ALL following ET in 1986 (10). In 1996, Berkahn et al. reported a case of T-ALL transformation from ET (11). Nagai et al. reported a case of Ph+ B-ALL that developed after a long duration of ET, and mutational analysis of JAK2 showed that the B-ALL clone did not originate from the ET clone with the JAK2-V617F mutation (12). A case of a patient with Ph+ALL, developed from MPL-mutated ET, was reported by Ma et al. in 2021. Molecular studies also indicated that the clone did not originate from the ET clone carrying the MPL p.(W515L) variant (13). In 2017, Hilal et al. reported a case of B-ALL with der(1)t(1;19)(p13;p13.1) arising in the setting of CALR exon 9-mutated ET (14). Medawar et al. reported a case with a history of JAK2/CALR-negative ET in which the patient subsequently developed T-ALL (15). Thus, including the case reported here, there have been seven reported cases of acute lymphoblastic leukemia transformation following ET (Table 1). The average age at which the patients developed ALL was 61 years (range: 49–87 years), with five cases transforming to B-ALL, two cases to Ph+ ALL, and two cases to T-ALL. The median time to progression was 11.4 years (range: 2 months–26 years). At the time of ET diagnosis, four patients underwent mutation testing (JAK2/CALR/MPL), revealing two triple-negative cases, one with a JAK2 mutation, and one with a CALR mutation. At the time of transformation, NGS was performed in three patients, identifying mutations in SETD2 (c.5126_5127insCC), BTK (splice site c.1408A > G), MPL p.(W515L), TET2 (R1261C), NOTCH1 (L1678P), and PHF6 (splice site c.374 + 1G > A).
The clonal origins of post-ET ALL remain controversial. In our case, the absence of shared driver mutations (JAK2/CALR/MPL in ET vs. SETD2/IKZF1 in ALL) supports clonal independence, consistent with prior reports of MPL- or JAK2-mutated ET progressing to Ph+ B-ALL (12, 13, 15). Notably, similar findings were observed in MPL-mutated ET transforming to Ph+ ALL, where the low VAF of MPL in leukemic blasts (2.59%) contrasted with the dominant BCR::ABL1 clone, further indicating divergent clonal evolution (13). A recent review of 43 Ph+ ALL cases corroborates that SETD2 mutations and IKZF1 deletions are hallmark alterations in Ph+ ALL but unrelated to MPN biology (17), reinforcing the “two-hit” model of random co-occurrence. From a mechanistic perspective, the coexistence of ET and ALL may reflect a permissive BM microenvironment. Experimental studies suggest that MPN remodels the endosteal BM niche into a self-reinforcing leukemic niche through thrombopoietin, chemokine ligand 3 (CCL3), and direct cell-cell interactions (16). In the case reported here, NGS suggested mutations in the SETD2 gene and deletion of the IKZF1 gene, which are common molecular alterations in Ph+ ALL. Although baseline NGS was unavailable at ET diagnosis, no common MPN mutations (SF3B1, U2AF1, TP53, IDH2, and EZH2) were detected at Ph+ ALL transformation (18), further supporting clonal divergence. Future studies using single-cell sequencing could elucidate whether pre-ET hematopoietic stem cells harbor latent lymphoid-primed mutations.
In the pre-tyrosine kinase inhibitor (TKI) era, Ph+ ALL was typically resistant to conventional chemotherapy, with allo-HSCT being the only curative option. The long-term survival rate was only 10%–20% and even worse for elderly or unfit patients (19). The advent of TKIs and immunotherapies has revolutionized management. For frail patients, TKI-based regimens with reduced-intensity chemotherapy or corticosteroids achieve high remission rates. The EWALL-PH-01 study reported 96% complete remission (CR) and 36% 5-year overall survival (OS) with dasatinib plus low-dose chemotherapy in 71 patients with Ph+ ALL, with a median age of 69 years. Furthermore, 65% of patients achieved a 3-log reduction in BCR::ABL1 transcript levels during consolidation (20). In 2020, Foà et al. used a dasatinib combined with a corticosteroid induction regimen followed by two cycles of blinatumomab to treat 63 patients with Ph+ ALL, achieving 18-month OS and disease-free survival (DFS) rates of 95% and 88%, respectively (21). Our patient had an atypical BCR::ABL1 fusion gene (e13a3). Whether such patients respond similarly to TKIs and CD3/CD19 bispecific antibodies as those with common BCR::ABL1 fusion genes is currently unreported. In chronic myeloid leukemia (CML) patients, the available literature suggests that e13a3/e14a3 transcripts correlate with superior imatinib responses (22, 23). The patient reported here experienced unstable angina upon admission. Acute leukemias, particularly with hyperleukocytosis, are associated with endothelial injury, hyperviscosity, and thromboinflammatory complications. These mechanisms may precipitate microvascular angina even in the absence of obstructive coronary artery disease (24, 25). Olverembatinib is a novel third-generation TKI approved in China (26). In a multi-center study, 20 patients with de novo Ph+ ALL were treated with olverembatinib-based regimens as frontline therapy. All patients achieved CR, and 85% achieved CMR within 3 months (27). Olverembatinib is chosen due to its superior safety profile and is prioritized over other TKIs in patients with cardiovascular comorbidities (26, 27). Our patient declined allo-HSCT, and blinatumomab was later introduced during consolidation to target residual disease, as supported by the EWALL-PH-01 trial (21). The patient achieved sustained molecular remission with olverembatinib and blinatumomab, avoiding intensive chemotherapy—a strategy aligned with emerging protocols for frail populations (21, 27). This contrasts with historical cases of ET-ALL treated with conventional regimens, where outcomes were poor (5, 10). While the patient currently refuses allo-HSCT, this remains a curative option for relapse, particularly with reduced-intensity conditioning (RIC) protocols (20). Additionally, emerging therapies such as CAR-T cells targeting CD19/CD22 may provide salvage options for refractory cases (28). In conclusion, this is a rare case of Ph+ ALL with e13a3 fusion transcripts following ET. It underscores the need for rigorous lineage evaluation in patients with ET with cytopenia or circulating blasts to exclude lymphoid transformation, which may mimic myelofibrosis progression. Molecular sequencing is important to elucidate whether pre-ET hematopoietic stem cells harbor latent lymphoid-primed mutations. Ph+ ALL with e13a3 transcripts may achieve durable remission with TKI-based regimens regardless of ET background. More data are needed to accumulate experience in treating Ph+ ALL following ET.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Babakhanlou R Masarova L Verstovsek S. A review of essential thrombocythemia and its complications. Clin Adv Hematol Oncol. (2023) 21:76–84.36780473 · pubmed ↗
- 2Tefferi A Guglielmelli P Larson DR Finke C Wassie EA Pieri L. Long-term survival and blast transformation in molecularly annotated essential thrombocythemia, polycythemia vera, and myelofibrosis. Blood. (2014) 124:2507–13; quiz 2615. doi: 10.1182/blood-2014-05-579136 25037629 PMC 4199952 · doi ↗ · pubmed ↗
- 3Godfrey AL Green AC Harrison CN. Essential thrombocythemia: challenges in clinical practice and future prospects. Blood. (2023) 141:1943–53. doi: 10.1182/blood.2022017625 36379024 · doi ↗ · pubmed ↗
- 4Guglielmelli P Vannucchi AM. Current management strategies for polycythemia vera and essential thrombocythemia. Blood Rev. (2020) 42:100714. doi: 10.1016/j.blre.2020.100714 32546373 · doi ↗ · pubmed ↗
- 5Campbell PJ Baxter EJ Beer PA Scott LM Bench AJ Huntly BJ. Mutation of JAK 2 in the myeloproliferative disorders: timing, clonality studies, cytogenetic associations, and role in leukemic transformation. Blood. (2006) 108:3548–55. doi: 10.1182/blood-2005-12-013748 16873677 · doi ↗ · pubmed ↗
- 6Mahdi D Spiers J Rampotas A Polverelli N Mc Lornan DP. Updates on accelerated and blast phase myeloproliferative neoplasms: Are we making progress? Br J Haematol. (2023) 203:169–81. doi: 10.1111/bjh.19010 37527977 · doi ↗ · pubmed ↗
- 7Tefferi A Barbui T. Polycythemia vera and essential thrombocythemia: 2021 update on diagnosis, risk-stratification and management. Am J Hematol. (2020) 95:1599–613. doi: 10.1002/ajh.26008 32974939 · doi ↗ · pubmed ↗
- 8Tefferi A Vannucchi AM Barbui T. Essential thrombocythemia: 2024 update on diagnosis, risk stratification, and management. Am J Hematol. (2024) 99:697–718. doi: 10.1002/ajh.27216 38269572 · doi ↗ · pubmed ↗