Midline defect with corpus callosum agenesis, vermian hypoplasia and median cleft lip palate
Clara Illi, Josefine Theresia Koenigbauer, Alexander Weichert

TL;DR
A case report describes a fetus with multiple midline brain and facial abnormalities, including corpus callosum agenesis and cleft palate, with no identified genetic cause.
Contribution
Presents a rare case of midline defects with no detectable genetic cause, highlighting the importance of prenatal imaging and genetic testing.
Findings
Midline brain abnormalities and facial deformities were detected prenatally but no genetic cause was identified.
Exome sequencing failed to reveal pathogenic or significant variants despite multiple anomalies.
The case highlights the value of prenatal findings in guiding clinical decisions.
Abstract
Midline defects in the brain may be related to genetic syndromes. Association with facial anomalies and skeletal deformities has been described. In the present case, a routine second trimester scan revealed cerebral abnormalities (corpus callosum agenesis, cerebellar cleft due to vermian hypoplasia, ventriculomegaly), suspected cortical developmental disorder, hypertelorism, a hypoplastic nasal bone, a small median cleft lip and palate, abnormal facial profile, as well as syndactyly of the left hand involving the fourth and fifth finger. Genetic testing revealed a normal karyotype. Subsequent trio exome sequencing did not identify any pathogenic variants or variants of unknown significance. The vaginal delivery at term and postnatal adaptation were uneventful. Postnatal neurosonographic imaging and clinical evaluation confirmed the prenatal findings. Both mother and child were…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
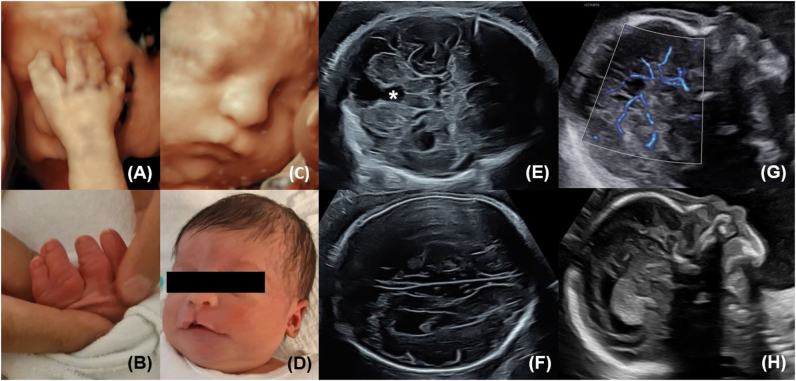 Figure 1
Figure 1Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsFetal and Pediatric Neurological Disorders · Prenatal Screening and Diagnostics · Cleft Lip and Palate Research
What is already known about this topic?
- –Midline defects are rare and may be associated with genetic syndromes.
What does this study add?
- –We report prenatal findings of a midline defect with corpus callosum agenesis, vermian hypoplasia and median cleft lip palate with a normal karyotype and no pathological finding in trio exome sequencing.
- –Genetic testing can help parents and healthcare providers with informed decision making regarding affected pregnancy. Still, some fetal anomalies may be unsolved.
Introduction
Prenatal diagnosis of fetal anomalies regularly confronts healthcare providers and parents with critical questions regarding further procedures and invasive diagnostic. Genetic testing can help with informed decision-making regarding affected pregnancy.
Case presentation
A 24-year-old woman, Gravida II Para I, presented at 21 + 6 gestational weeks for a routine second anomaly trimester scan. The fetal phenotype included a female fetus with corpus callosum agenesis, cerebellar cleft due to vermian hypoplasia, ventriculomegaly, suspected cortical developmental disorder, hypertelorism, a small median cleft lip and palate, as well as syndactyly of the left hand involving the fourth and fifth finger (Figure 1). Fetal and maternal Doppler indices were normal. After genetic counseling the parents opted for further genetic testing and an amniocentesis was performed resulting in a normal karyotype. Trio exome sequencing did not reveal any pathogenic variant or variant of unknown significance. Trio exome sequencing is a method of sequencing the exomes of the affected individual and both biological parents to identify potential disease-causing variants. By comparing the exomes of the affected individual with those of the parents, it helps pinpoint inherited genetic mutations and de novo variants that may contribute to the condition. The family received intensive consultations with pediatric neurologists. A fetal MRI was offered but not favored by the parents. Regular growth scans were normal. The child was delivered vaginally at term without any complications. The sonographic findings were confirmed postnatally. In addition, short fingers were noted (Figure 1). The postnatal adaption was physiological except for some feeding problems. Child and mother could be discharged home after one day.
Fetal phenotype: 3D images (A, C), B-mode images (E–H), postnatal images (B, D). (A, B) Syndactyly of the left hand involving fourth and fifth finger; short fingers. (C, D) Small median cleft lip and palate. (E) Cerebellar cleft due to vermian hypoplasia (). (F–H) Corpus callosum agenesis.*
Discussion
If the above-mentioned fetal phenotype is detected on prenatal ultrasound and standard cytogenomic testing is non-diagnostic, trio exome sequencing is indicated.
The differential diagnoses include Hartsfield-Bixler-Demyer Syndrome, Oro-Facial-Digital-Syndromes, Ectrodactyly Ectodermal Dysplasia Cleft Lip/Palate Syndrome and Acrocallosal Syndrome. All mentioned syndromes present a wide range of clinical variability.
Hartsfield-Bixler-Demyer Syndrome, also known as fibroblast growth factor 1 (FGFR1)-related Hartsfield Syndrome, is a rare genetic disorder characterized by a combination of holoprosencephaly and ectrodactyly spectrum disorder, as well as facial anomalies including cleft lip and palate [1]. Other associated midline brain malformations include corpus callosum agenesis, absent septum pellucidum and vermian hypoplasia.
The Oro-Facial-Digital Syndromes include at least 13 genetically heterogeneous disorders characterized by anomalies of the oral cavity, facial features and digits. Additional abnormalities in the brain and visceral organs have been described [2].
Ectrodactyly-Ectodermal Dysplasia-Cleft Lip/Palate Syndrome is a rare genetic disorder characterized by ectrodactyly, ectodermal dysplasia and cleft lip/palate. It is an autosomal dominant condition primarily associated with variations in the tumor protein 63 (TP63) gene [3].
Acrocallosal Syndrome is an autosomal recessive disorder caused by variants in the kinesin family member 7 (KIF7) gene. It is characterized by agenesis of the corpus callosum, midline brain and facial abnormalities, polydactyly, and in most cases psychomotor developmental delay [4].
Corpus callosum agenesis is one of the most frequent brain malformations and involves congenital absence of all or parts of the corpus callosum. It appears as an isolated condition or in association with a genetic syndrome. A genetic cause can be identified in 12.5 % with chromosomal microarray and up to 47 % with whole exome sequencing [5]. The postnatal outcome varies considerably, even in isolated corpus callosum agenesis outcomes may range from normal in 65 % to mild to severe neurodevelopmental impairments in 35 % [6].
The present case shows that even highly pathologic sonographic findings are not necessarily associated with pathologic genetic findings in trio exome sequencing or a specific genetic syndrome.
In conclusion, genetic as well as specific pediatric counseling is recommended when anomalies are suspected to discuss the nature of the disorder, inheritance patterns, and implications for the pregnancy and family planning.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Nicolas S Isabelle M Nelle L Camille P Deepthi CS Boyan D FGFR 1 mutations cause Hartsfield syndrome, the unique association of holoprosencephaly and ectrodactyly J Med Genet 20135058510.1136/jmedgenet-2013-10160323812909 PMC 3756455 · doi ↗ · pubmed ↗
- 2Franco B Thauvin-Robinet C Update on oral-facial-digital syndromes (OFDS)Cilia 201651210.1186/s 13630-016-0034-427141300 PMC 4852435 · doi ↗ · pubmed ↗
- 3Koul M Dwivedi R Upadhyay V Ectrodactyly-ectodermal dysplasia clefting syndrome (EEC syndrome)J Oral Biol Craniofac Res 20144135910.1016/j.jobcr.2014.08.00225737931 PMC 4252378 · doi ↗ · pubmed ↗
- 4Putoux A Thomas S Coene KL Davis EE Alanay Y Ogur G KIF 7 mutations cause fetal hydrolethalus and acrocallosal syndromes Nat Genet 201143601610.1038/ng.82621552264 PMC 3674836 · doi ↗ · pubmed ↗
- 5Tsai P Shinar S Agenesis of the corpus callosum: what to tell expecting parents?Prenat Diagn 20234315273510.1002/pd.644737794643 · doi ↗ · pubmed ↗
- 6D’Antonio F Pagani G Familiari A Khalil A Sagies TL Malinger G Outcomes associated with isolated agenesis of the corpus callosum: a meta-analysis Pediatrics 201613810.1542/peds.2016-044527581855 · doi ↗ · pubmed ↗