Navigating the complexity of atypical teratoid/rhabdoid tumor (ATRT) in pediatric neuro-oncology: Insights from clinical spectrum to therapeutic challenges
Ali Msheik, Mohamad Yazbeck, Abdulla Illeyan, Youssef Comair

TL;DR
This paper presents a case of a rare and aggressive pediatric brain tumor, showing that early diagnosis and aggressive treatment can lead to long-term remission.
Contribution
The paper contributes a detailed case study demonstrating the effectiveness of multimodal therapy in managing atypical teratoid/rhabdoid tumor (ATRT).
Findings
Early diagnosis and aggressive multimodal therapy led to no recurrence in a 2-year-old ATRT patient.
Maximal safe resection followed by chemotherapy is critical for improving long-term outcomes in ATRT.
MRI features like heterogeneous enhancement and solid-cystic lesions are typical for ATRT.
Abstract
Atypical teratoid/rhabdoid tumor (ATRT) is a rare and aggressive pediatric central nervous system malignancy, accounting for only 1–2 % of cases. Primarily affecting children under three years old, ATRT poses significant diagnostic and therapeutic challenges, with high recurrence rates and poor prognosis due to its rapid progression and lack of a standardized treatment protocol. We report the case of a 2-year-old male diagnosed with infratentorial ATRT after presenting with abnormal gait, vomiting, and ataxia following minor head trauma. Magnetic resonance imaging (MRI) revealed a mixed solid-cystic cerebellar lesion, prompting surgical resection. Despite postoperative chemotherapy, tumor progression was noted, leading to a second craniotomy, which achieved complete resection. Serial follow-up MRI until February 2025 showed no evidence of recurrence, and the patient remains…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
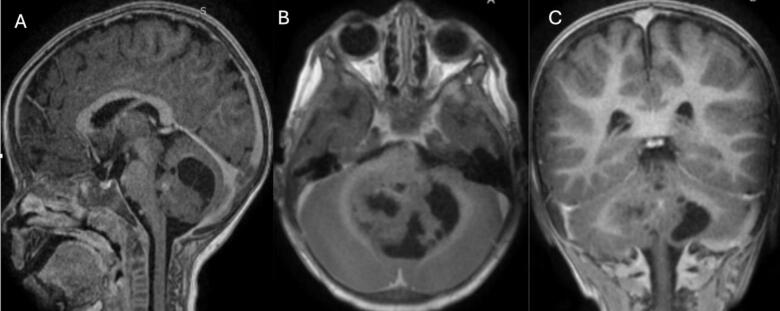 Figure 1
Figure 1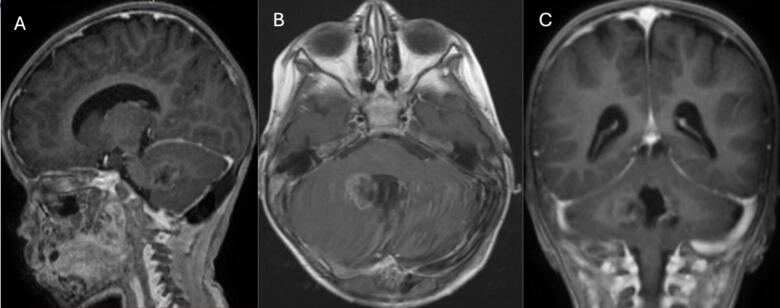 Figure 2
Figure 2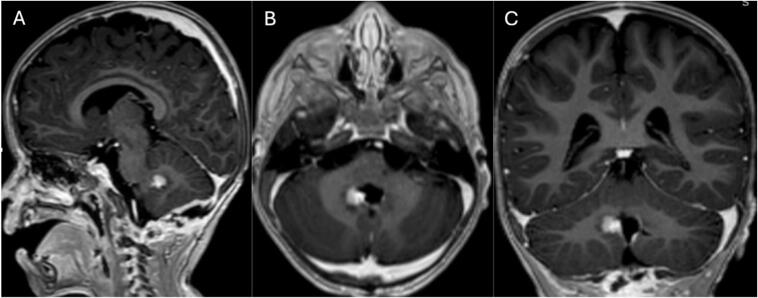 Figure 3
Figure 3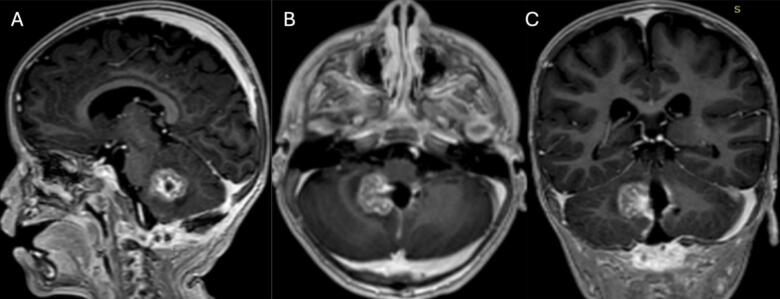 Figure 4
Figure 4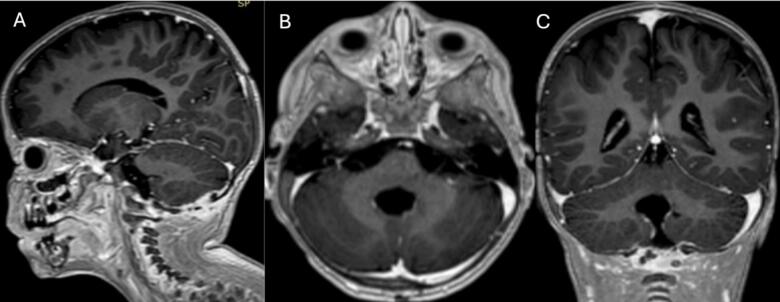 Figure 5
Figure 5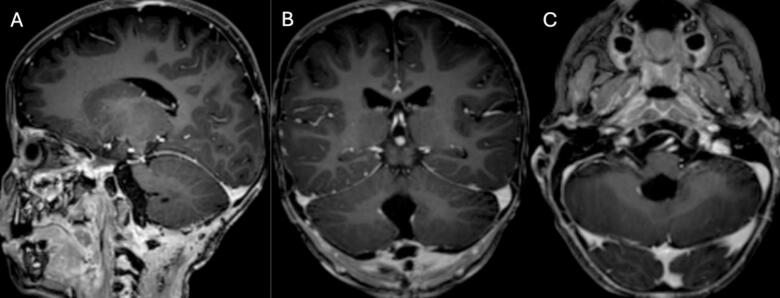 Figure 6
Figure 6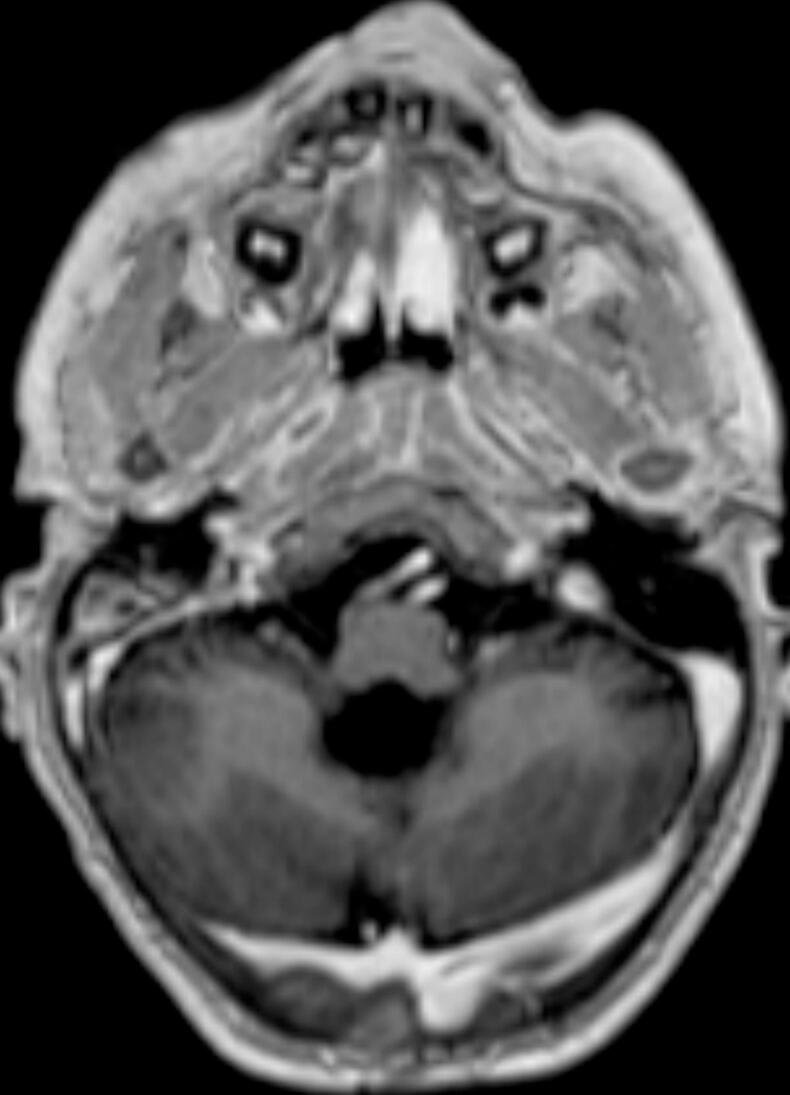 Figure 7
Figure 7Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsChromatin Remodeling and Cancer · Gestational Trophoblastic Disease Studies · Cancer Mechanisms and Therapy
Introduction
1
Atypical teratoid/rhabdoid tumor (ATRT) constitutes 1–2 % of pediatric central nervous system (CNS) neoplasms [1] and ranks as the predominant malignancy in children under 36 months at 17.3 % [1]. ATRT is highly aggressive. It is infratentorial in half of the reported cases [2]. A pediatric cohort reported a 34 % supratentorial incidence of ATRT [2]. While metastasis is observed in only 35.3 % of patients older than 6 months [2], 100 % of the patients under 6 months showed metastasis at diagnosis [3]. In adults, the sellar region and cerebral hemispheres are commonly affected. ATRTs histologically feature rhabdoid cells and mixed components of neuroectodermal, ectodermal, and mesenchymal cells. As per the World Health Organization (WHO) classification, embryonal tumors are designated as Grade 4 tumors [4] except CNS tumors with BCL6 corepressor internal tandem duplication and cribriform neuroepithelial tumors [4].
Cerebellar tumors manifest as headache, vomiting, gait abnormalities, and instability. The tumor appears typically hyperintense on T2-weighted magnetic resonance imaging (MRI), images, except for hemorrhagic regions, and exhibits heterogeneously enhancing T1 images with contrast [5]. Immunohistochemistry and genetic testing are for definitive diagnosis because the rhabdoid cells, which are characteristic, constitute a limited portion of the specimen. Up to 95 % of ATRTs display two inactive copies of the integrase interfactor-1 (INI-1) gene. Rhabdoid cells commonly express epithelial membrane antigen (EMA), vimentin, and smooth muscle actin (SMA) [6].
The prognosis is exceptionally poor due to recurrence at the primary tumor site or distant metastases in blood and cerebrospinal fluid (CSF) and the lack of standard treatment protocol for ATRTs. The mean survival despite surgical intervention, radiotherapy, and high-dose chemotherapy (HDCT) is 6–18 months [7]. Disease progression (PD) is observed in 77 % of cases, typically within a median of 160 days post-diagnosis [8]. Some authors suggest improved survival with the absence of metastasis at diagnosis, early adjuvant radiotherapy and HDCT, and diagnoses made after the year 2011 [8].
This report describes the case of a 2-year-old male child diagnosed with an infratentorial tumor, which underwent surgical intervention and was subsequently identified as an Atypical Teratoid Rhabdoid Tumor (ATRT).
Case presentation
2
This is a case of a 3-year-old pediatric male patient born on December 13, 2018. At the age of 2, he manifested abnormal gait, vomiting, and ataxia, following a minor head trauma. An MRI examination showed a mixed solid cystic lesion measuring 56 × 37 × 36 mm within the cerebellum (Figs. 1 A, B, and C). Laboratory workup was unremarkable.Fig. 1. Magnetic resonance imaging of the brain: First MRI done before the first.Fig. 1
The patient underwent craniotomy and surgical excision in January 2021. A post-procedure MRI revealed a residual enhancing lesion and the histopathological analysis confirmed the presence of an ATRT (Figs. 2 A, B, and C). ACNS0333 chemotherapy regimen was initiated. A second MRI on February 5, 2021, revealed an increase in the size of the enhancing residual lesion (Figs. 3 A, B, and C). A second craniotomy was done on April 16, 2021, with complete intraoperative resection. The patient was followed with interval MRI until February 2025 (Fig. 4, Fig. 5, Fig. 6, Fig. 7). The patient is symptom-free with no radiological evidence of recurrence.Fig. 2. Magnetic resonance imaging of the brain: First MRI done after the first surgical intervention.Fig. 2. Fig. 3Magnetic resonance imaging of the brain: first imaging after chemotherapy initiation post-surgical intervention.Fig. 3. Fig. 4Magnetic resonance imaging of the brain: Second MRI after the First craniotomy showing an increase in the size of the enhancing residual lesion.Fig. 4. Fig. 5Magnetic resonance imaging of the brain: First imaging after the second craniotomy showing complete resection.Fig. 5. Fig. 6Magnetic resonance imaging of the brain: Second follow-up imaging showing total remission.Fig. 6. Fig. 7Axial cut of a T1 MRI seqquence done on Febraury 2025. There is no evidence of radiological.Fig. 7
Discussion
3
Notably, the age-dependent disparity in metastatic disease underscores the distinct pathological characteristics in infants, with all those under six months displaying metastatic involvement, compared to 35.3 % in older infants [[9], [10], [11], [12], [13], [14]]. The workup for primary lesions and metastasis was negative. No pathology was detected on a whole-body CT scan. All tumor markers were negative. The definitive diagnosis necessitates immunohistochemistry and genetic testing due to the limited representation of rhabdoid cells in the specimen. The commonality of inactivation of both copies of the integrase interfactor-1 (INI-1) gene and the expression of epithelial membrane antigen (EMA), vimentin, and smooth muscle actin (SMA) in rhabdoid cells further underlines the molecular intricacies of ATRT.
Histologically akin to medulloblastomas, ATRTs demonstrate an embryonic origin, featuring rhabdoid cells and a composite structure comprising neuroectodermal, ectodermal, and mesenchymal elements, classifying them as Grade 4 tumors according to the World Health Organization (WHO) criteria. The diagnostic process involves magnetic resonance imaging (MRI), revealing distinct radiological features such as hyperintensity on T2-weighted images and heterogeneous enhancement on T1 images with contrast, mirroring similarities with medulloblastomas. The imaging findings in this patient align with the literature. ATRTs typically present as mixed solid-cystic lesions, with hyperintensity on T2-weighted images and heterogeneous enhancement on T1 images with contrast. The MRI findings in this case showed a mixed solid-cystic lesion, which is consistent with the known radiological characteristics of ATRTs. While ATRTs and medulloblastomas share some imaging characteristics, such as hyperintensity on T2-weighted images and contrast enhancement on T1, ATRTs often exhibit more heterogeneous enhancement due to their mixed cellular composition. Medulloblastomas, on the other hand, are usually more homogeneously enhancing and less likely to be cystic. The imaging features in this patient suggest similarities but also highlight key differences.
Despite the preoperative differential diagnosis, surgery is a key component of treatment for both ATRTs and medulloblastomas. In both cases, maximal safe resection improves prognosis by reducing tumor burden before adjuvant therapy. Therefore, even if the diagnosis were medulloblastoma, surgery would still have been beneficial. However, the management would have differed primarily in the choice of adjuvant therapy. ATRTs require an aggressive multimodal approach, including intensive chemotherapy (e.g., ACNS0333 regimen) and often radiation, depending on the patient’s age. Medulloblastomas are typically managed with surgical resection followed by risk-stratified treatment, including craniospinal irradiation and chemotherapy, which varies depending on molecular subtypes. In this case, if the lesion had been a medulloblastoma, treatment might have included craniospinal irradiation earlier, given the high risk of CSF dissemination in medulloblastomas. However, since ATRTs are more aggressive, the chosen management approach for this patient was appropriate.
Despite advancements in therapeutic modalities, a standardized treatment protocol for ATRTs remains elusive, contributing to a particularly grim prognosis with a mean survival of 6–18 months. The challenges are further underscored by the observed recurrence at the primary tumor site and the occurrence of distant metastases in blood and cerebrospinal fluid (CSF), both major contributors to patient mortality. The discussion delves into the complexities of disease progression (PD), documented in 77 % of cases within a median of 160 days post-diagnosis. Moreover, a nuanced exploration of the literature reveals potential correlations between improved survival and various factors, including the absence of metastasis at diagnosis, early adjuvant radiotherapy and high-dose chemotherapy (HDCT), and diagnoses made after the year 2011, indicative of evolving therapeutic paradigms [[15], [16], [17], [18], [19], [20], [21]].
The summarized studies in Table 1 highlight the challenges and variability in ATRT presentation, treatment, and outcomes [[22], [23], [24]]. The Children’s Oncology Group (COG) ACNS0333 study, with 64 cases, underscores the aggressive nature of ATRT and the necessity of intensive chemotherapy and surgical intervention, reporting a 4-year overall survival (OS) of 54 % for infratentorial cases. Similarly, the case report by Paun et al. describes a posterior fossa ATRT managed with a two-stage surgical approach and chemotherapy, leading to a positive outcome. The case series in Frontiers in Surgery further emphasizes the importance of multimodal therapy in achieving a better prognosis. Compared to these studies, our case aligns with the literature in terms of presentation—manifesting with ataxia and gait disturbances—and radiological characteristics, showing a solid cystic lesion in the posterior fossa. However, despite an initial recurrence, our patient demonstrated long-term survival without recurrence, suggesting that early aggressive surgical intervention combined with ACNS0333-based chemotherapy may improve outcomes in select pediatric ATRT cases.Table 1. Summary of selected studies reporting treatment approaches and outcomes in infants and young children with Atypical teratoid/rhabdoid tumors (ATRTs), highlighting tumor location, treatment modalities, and survival outcomes.Table 1. StudyNumber of casesAge rangeTumor locationTreatmentOutcomeReddy et al., [22]64< 3 yearsInfratentorial and SupratentorialSurgery + Intensive Chemotherapy ± Radiation4-year OS: 54 % (infratentorial), 35 % (supratentorial)Paul et al. [23]19 monthsPosterior FossaTwo-stage surgery + ChemotherapyPositive outcome, survival beyond standard prognosisGou et al. [24]1216–30 monthsInfratentorial and SupratentorialSurgery + Chemotherapy ± RadiationHighlighted multimodal therapy benefits
Conclusion
4
This case highlights the complexities of managing pediatric ATRT, emphasizing the importance of a multidisciplinary approach. The imaging findings were consistent with ATRTs, showing similarities to medulloblastomas but with distinct characteristics. Surgery played a crucial role in tumor control, and while management would have differed for medulloblastoma—particularly with earlier craniospinal irradiation—the aggressive multimodal approach used here was essential for ATRT. This case underscores the challenges in achieving complete tumor eradication and the necessity of close radiological and clinical follow-up to optimize patient outcomes.
Consent
Consent was taken from the mother of the patient because he is a pediatric patient. The consent form is signed and available upon request.
Ethical approval
The study was exempted from ethical approval as the information was reported anonymously.
Disclaimer
This article was reported according to the SCARE criteria [25].
Sources of funding
This study was not funded.
Declaration of competing interest
The authors declare no conflict of interest.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Park M.Han J.W.Hahn S.M.Atypical teratoid/rhabdoid tumor of the central nervous system in children under the age of 3 years Cancer Res. Treat.53202137838810.4143/crt.2020.75633138347 PMC 8053862 · doi ↗ · pubmed ↗
- 2Hasselblatt M, Kordes U, Wolff J, et al.: Abstracts of the 15th international symposium on pediatric neuro-oncology. Pediatric Neuro-Oncology. June 24-27. 2012, 14:10.1093/neuonc/nos 091.PMC 348333722745951 · doi ↗ · pubmed ↗
- 3Heath M.Jaimes N.Lemos B.Clinical characteristics of Merkel cell carcinoma at diagnosis in 195 patients: the AEIOU features J. Am. Acad. Dermatol.58200837538110.1016/j.jaad.2007.11.02018280333 PMC 2335370 · doi ↗ · pubmed ↗
- 4Huisman T.A.Tumor-like lesions of the brain Cancer Imaging 9 Spec No A(Special issue A):S 10-32009 Oct 210.1102/1470-7330.2009.9003 PMID: 19965288; PMCID: PMC 2797474 PMC 279747419965288 · doi ↗ · pubmed ↗
- 5Han E.Kim J.Jung M.J.Chin S.Lee S.W.Moon A.Malignant rhabdoid tumor of the kidney in an adult with loss of INI 1 expression and mutation in the SMARCB 1 gene J. Pathol. Transl. Med.55202114515310.4132/jptm.2021.01.2633677955 PMC 7987524 · doi ↗ · pubmed ↗
- 6Albain K.S.Swann R.S.Rusch V.W.Radiotherapy plus chemotherapy with or without surgical resection for stage III non-small-cell lung cancer: a phase III randomised controlled trial Lancet 374200937938610.1016/S 0140-6736(09)60737-619632716 PMC 4407808 · doi ↗ · pubmed ↗
- 7Nesvick C.L.Nageswara Rao A.A.Raghunathan A.Biegel J.A.Daniels D.J.Case- based review: atypical teratoid/rhabdoid tumor Neurooncol. Pract.6201916317810.1093/nop/npy 03731386032 PMC 6656328 · doi ↗ · pubmed ↗
- 8Baiano C.Della Monica R.Franca R.A.Atypical teratoid rhabdoid tumor: a possible oriented female pathology?Front. Oncol.12202210.3389/fonc.2022.854437 PMC 901082435433419 · doi ↗ · pubmed ↗