Synthesis and Application of PN-Supported Mn(I) Carbonyl Alkyl Complexes
Claudia Rabijasz, Stefan Weber, Berthold Stöger, Karl Kirchner

TL;DR
Scientists created and tested new manganese complexes for use in chemical reactions, finding some to be effective catalysts.
Contribution
New Mn(I) carbonyl alkyl complexes with PN ligands were synthesized and shown to catalyze specific reactions efficiently.
Findings
fac-[Mn(PCyNMe)(CO)3Me] is effective for phenylacetylene dimerization.
fac-[Mn(PPhNMe)(CO)3Me] catalyzes the hydroboration of 4-chlorostyrene efficiently.
Abstract
This work comprises the synthesis and characterization of aminophosphine (PN)-derived Mn(I) carbonyl complexes and the preliminary investigation of their alkylated congeners for catalytic applications. The complexes fac-[Mn(PN)(CO)3Br] are obtained from the reaction of Mn(CO)5Br with the bidentate ligand PN = R2N(CH2)2PR′2, where R = Me, Et, and pyrrolidine and R′ = Ph, iPr, and Cy. Treatment of fac-[Mn(PN)(CO)3Br] with AgOTf yields fac-[Mn(PN)(CO)3OTf]. Upon reaction of fac-[Mn(PN)(CO)3OTf] with MeLi (R′ = alkyl) or MeMgCl (R′ = aryl), fac-[Mn(PN)(CO)3Me] is formed. fac-[Mn(PCyNMe)(CO)3Me] and fac-[Mn(PPhNMe)(CO)3Me] are identified as the best catalysts for the dimerization of phenylacetylene and the hydroboration of 4-chlorostyrene, respectively.
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
 Scheme 1
Scheme 1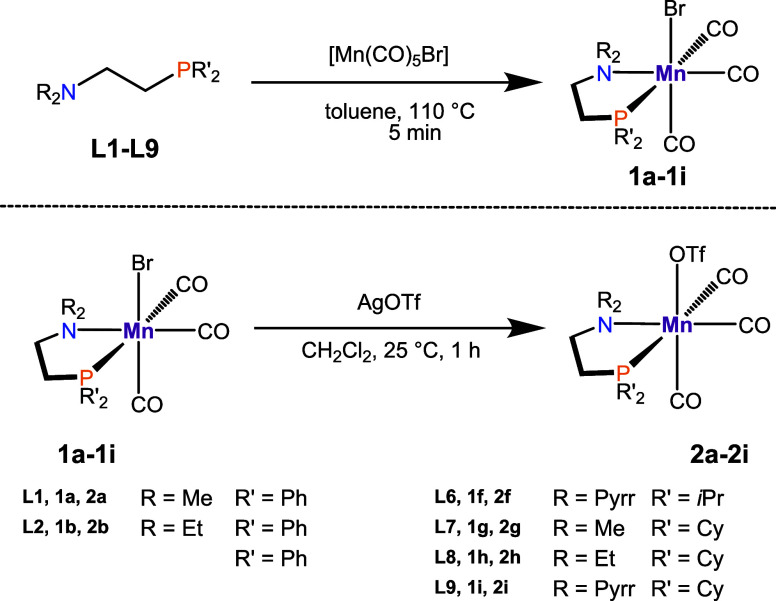 Scheme 2
Scheme 2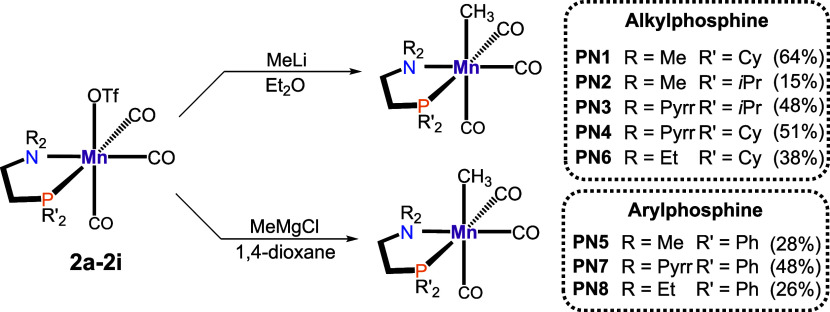 Scheme 3
Scheme 3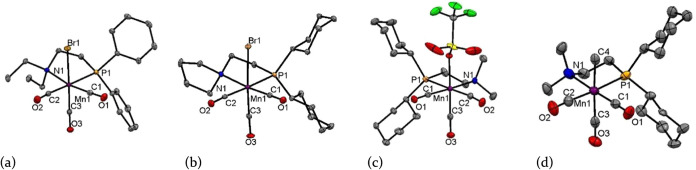 Figure 1
Figure 1 Scheme 4
Scheme 4| Mn1–P1 | 2.323 | 2.342 | 2.342 | 2.316 |
| Mn1–N1 | 2.239 | 2.186 | 2.189 | 2.210 |
| P1–Mn1–N1 | 82.17(3) | 84.26(4) | 83.84(4) | 83.65(7) |
| catalyst | conversion [%] | |
|---|---|---|
| 75 | 95:5 | |
| 35 | 96:4 | |
| 14 | 90:10 | |
| 12 | 90:10 | |
| n.d. | n.d. | |
| n.d. | n.d. | |
| trace | n.d. | |
| n.d. | n.d. | |
| >99 | 96:4 | |
| 67 | 79:21 | |
| 36 | 91:9 | |
| n.d | n.d. | |
| none | n.d | n.d. |
| catalyst | conversion [%] | A/B |
|---|---|---|
| 41 | >99:1 | |
| 67 | >99:1 | |
| 22 | >99:1 | |
| 16 | 97:3 | |
| 99 | >99:1 | |
| 93 | >99:1 | |
| 43 | 99:1 | |
| 57 | >99:1 | |
| >99 | 97:3 | |
| 80 | >99:1 | |
| 87 | >99:1 | |
| none | 6 | >99:1 |
- —Austrian Science Fund10.13039/501100002428
- —Austrian Science Fund10.13039/501100002428
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsAsymmetric Hydrogenation and Catalysis · Organometallic Complex Synthesis and Catalysis · Carbon dioxide utilization in catalysis
Introduction
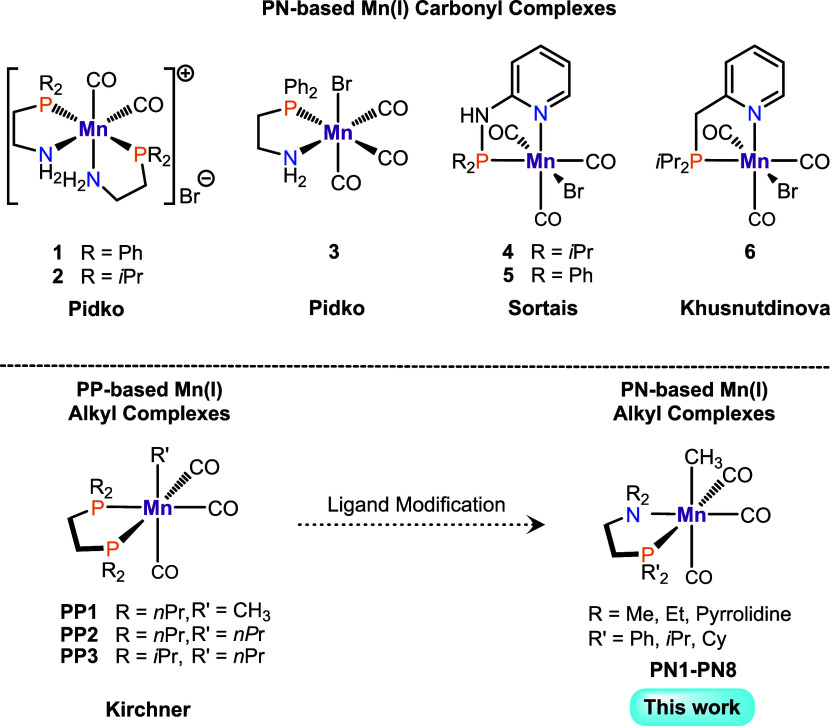
Organometallic catalysis plays an important role in industrial processes and promotes ongoing improvements in them.^1−4^ Driven by sustainability, replacement of noble metals by more abundant and less expensive counterparts attracted significant attention in recent years.^5^ Manganese, as the third most abundant transition metal in the Earth’s crust, emerged as a formidable player in homogeneous catalysis. Although pincer ligands are predominant in Mn(I)-catalyzed reactions,^6−9^ bidentate-based catalysts are encountered in several hydrogenation and hydrofunctionalization reactions. Selected examples for aminophosphine-based (PN) systems are depicted in Scheme 1 (1–6, top).^10−12^ All of the complexes described above were shown to operate via metal–ligand cooperation (MLC). In contrast, our group reported the bisphosphine (PP)-based Mn(I) complex fac-[Mn(nPr_2_PCH_2_CH_2_PnPr_2_)(CO)_3_Br] being capable of reduction of nitriles and ketones.^13^ By exchanging the bromide ligand with a methyl ligand to give the congener PP1 (Scheme 1, bottom), no additives were required for the hydrogenation of nitiriles.^14^ Moreover, the modified complexes PP2 and PP3 (Scheme 1, bottom) exhibited good to excellent yields in various hydrogenation reactions.^15,16^ Other transformations, e.g., dimerization of alkynes,^17^ dehydrogenative silylation, and hydroboration of terminal alkenes were also catalyzed by PP3.^18,19^ Since MLC is not possible in these systems, the reactivity of these complexes is based on inner-sphere pathways. These can be enabled by migratory insertion of the alkyl group into the adjacent CO ligand, followed by protonation of the formed acyl and dissociation of the aldehyde.^20−23^
Representative Mn(I) Complexes Supported by PN Ligands
However, the ligand design proved to be crucial to achieving high reactivity of the active species. We attribute this to the migratory aptitude of the alkyl group since this step was found to be rate-limiting in all studied transformations. To extend this concept, we were interested in a mixed donor set to facilitate migratory insertion due to a different donor trans to the carbonyl ligands. Thus, we decided to investigate PN-based systems. The combination of a hard nitrogen and a soft phosphorus donor is a distinctive feature of PN ligands. Furthermore, this can enable the dissociation of the nitrogen donor, thus generating a vacant coordination site for substrate binding.^24^ While PN-based systems were proven to enhance catalytic activity in various transformations,^25−29^ investigations on PN-based manganese reactions are scarce.^30,31^
Herein, we report the synthesis and characterization of a series of PN-supported Mn(I) tricarbonyl triflate complexes as precursors for the corresponding methyl complexes. These complexes were obtained by ligation of aminophosphines with [Mn(CO)_5_Br], followed by treatment with AgOTf. A procedure for alkylation was developed to give the methyl complexes. In order to gain insight into the catalytic activity of these alkyl complexes, preliminary investigations were conducted.
Results and Discussion
Synthesis and Characterization
Upon heating PN ligands with [Mn(CO)_5_Br], 1a–1i could be synthesized in a short reaction time (Scheme 2, top). All bromide species were obtained as yellow or orange powders in a 23–91% isolated yield.
Complexation of PN Ligands with [Mn(CO)5Br] (Top) and Synthesis of Triflate Complexes 2 (Bottom)
Initial attempts of alkylation of complex 1 species were carried out through reduction with Na sand, similar to the successful syntheses of PP-related systems,^14^ followed by the addition of MeI or 1-bromopropane. In the case of MeI, undesired tetracarbonyl side products were formed, whereby the formation of a hydride species when utilizing 1-bromopropane was observed. Next, the treatment of complex 1 with carbon-based nucleophiles was investigated. However, neither direct alkylation utilizing MeLi nor MeMgCl was successful.^32,33^ The reaction of 1 with AgBF_4_ followed by the subsequent addition of ZnMe_2_ or ZnEt_2_ failed as well. Thus, the method was adopted by utilizing AgOTf.^34,35^ The transformation was achieved by mixing a solution of complex 1 in CH_2_Cl_2_ with AgOTf (1.50 equiv) (Scheme 2, bottom). The triflate congeners 2a–2i were obtained as yellow or orange powders, yielding 61–91%. Notably, 1 and 2 are air-stable compounds, but they are moderately light-sensitive. Regarding the ^31^P{^1^H} NMR spectra, all triflate complexes are slightly shifted downfield compared to their bromide analogues. The infrared (IR) spectra of complexes 1 and 2 display three distinctive CO signals in the carbonyl range of 2031–1874 cm^–1^, indicating a fac arrangement within an octahedral geometry.
The alkylation of 2 utilizing ZnMe_2_ or ZnEt_2_ resulted in undesired byproducts, similar to the alkylation of the bromide congeners. Treatment of 2 with n-BuLi was even less successful; in fact, a coordinated butyl group has never been observed. However, the alkylation of 2 was possible following two different procedures (Scheme 3). For complexes containing alkyl-substituted phosphines, methylation was achieved upon treatment with MeLi at −78 °C. In the case of aryl-substituted phosphines, the alkylation was carried with MeMgCl in the presence of 1,4-dioxane. These two procedures allowed the isolation of PN1–PN8 as yellow powders in moderate yields.
Synthesis of Methyl Complexes PN1–PN8 via Lithium or Grignard Reagents
The alkylated Mn(I) carbonyl compounds (PN1–PN8) were identified by ^1^H, ^31^P{^1^H}, and ^13^C{^1^H} NMR and IR spectroscopy as well as HR-MS (see Supporting Information). Significant downfield-shifted ^31^P{^1^H} NMR signals compared to the triflate species were observed. The characteristic methyl group, being bonded to the manganese center, exhibits a doublet ranging between 0.03 and −0.79 ppm in the ^1^H NMR spectra. The ^13^C{^1^H} NMR resonances of alkylphosphine-based compounds appear at ca. −4 ppm and those of phenylphosphine-based compounds at ca. −1 ppm. As a result of the stronger π-back-donation, the IR signals suggest the strongest Mn–CO bond for the methylated compounds, evidenced by the notably lower wavenumbers observed for PN, in contrast to 1 and 2.
Gratifyingly, we could also apply the introduced procedure for the synthesis of the bisphosphine-based complex fac-[Mn(P^Cy^P^Cy^)(CO)_3_Me] (PP4). This represents a more convenient protocol rather than reduction by Na/K followed by addition of MeI as reported for the synthesis of the similar complex PP1.^14^
Structure and Bonding
X-ray analysis of 1b, 1i, and 2g verified a slightly distorted octahedral geometry with fac-arranged CO ligands. The comparison of 1a(36) and 1b [Figure 1, (a)] highlights an interesting aspect; the Mn1–N1 distance is notably shorter in 1a (2.204 Å) compared to 1b (2.239 Å), while the Mn1–P1 distances are very similar (1a: 2.321 Å, 1b: 2.323 Å).
Structural views of (a) fac-[Mn(PPhNEt)(CO)3Br] (1b), (b) fac-[Mn(PCyNPyr)(CO)3Br] (1i), (c) fac-[Mn(PCyNMe)(CO)3OTf] (2g), and (d) fac-[Mn(PCyNMe)(CO)3Me] (PN1) showing 50% ellipsoids (H atoms are omitted for clarity; for selected bond lengths (Å) and bond angles (deg), see the Supporting Information).
Taking into account the related complex fac-[Mn(P^Ph^N^H^)(CO)3_Br] as reported by Pidko et al.,^10^ one can assert that the bond distance between manganese and nitrogen increases in the order −NH_2, −NMe_2_, and −NEt_2_. In this context, it is worth noting that the Mn1–N1 bond length in complex 1i [Figure 1, (b)], featuring a pyrrolidine scaffold, is 2.186 Å. This falls within the range of bond distances between fac-[Mn(P^Ph^N^H^)(CO)_3_Br] and fac-[Mn(P^Ph^N^Me^)(CO)3_Br] (1a). Fortunately, we were able to confirm the presence of one triflate species 2g using X-ray diffraction [Figure 1, (c)]. It provides a slightly distorted octahedral geometry with bond angles of 176.76(6)° (P1–Mn1–C2), 178.33(6)° (O4–Mn1–C3), and 176.12(6)° (N1–Mn1–C1). The smallest bite angle for aminophosphine of 82.17(3)° was observed in 1b and the largest angle of 84.26(4)° in 1i, bearing alkyl substituents on both the phosphorus and nitrogen atoms. In between were 1a and 2g with angles of 83.54(4)° and 83.84(4)°. A similar bite angle is known from the PP-supported complex where fac-[Mn(nPr_2_PCH_2_CH_2_PnPr_2)(CO)_3_Br] displays an angle of 83.56(4)°.^13^ Finally, the alkyl complex PN1 was confirmed via X-ray diffraction. A structural view is shown in Figure 1, (d). When comparing the crystal structures of 2g and PN1, a noticeable shortening of the Mn and basal CO distances is observed in PN1. These Mn—CO bond distances are 1.767 Å (Mn1–C1) and 1.801 Å (Mn1–C2) in PN1, while they are 1.811 Å (Mn1–C1) and 1.849 Å (Mn1–C2) in 2g, respectively. The apical CO distance to the Mn center is shorter in 2g at 1.780 Å (Mn1–C3) than in PN1 at 1.881 Å (Mn1–C3). This could be attributed to a stronger trans influence of the methyl group than that of the triflate group. The Mn–alkyl distance was 2.065 Å (Mn1–C20). The PN bite angle is nearly the same in both structures, 83.84(4)° in 2g and 83.65(7)° in PN1. A slightly larger bite angle of 85.53(2)° can be observed for the alkylated species PP3.^15^ Selected bond distances and bite angles are presented in Table 1.
Table 1: Selected Bond Distances (Å) and the Bite Angle (°) of 1b, 1i, 2g, and PN1
Catalytic Applications
At last, we were interested in the catalytic performance of PN-supported Mn(I) alkyl carbonyl complexes in hydrofunctionalization reactions. Preliminary investigations focused on the dimerization of phenylacetylene and the hydroboration of 4-chlorostyrene.
Dimerization of Phenylacetylene
PN1–PN8, PP4, and PP5(37) (Scheme 4) were applied in the dimerization of phenylacetylene; the results are summarized in Table 2 and compared with previously reported bisphosphine-based catalysts.
Investigated Mn(I) Methyl Complexes as Catalysts in This Study
Table 2: Catalytic Performance of PN- and PP-Based Mn(I) Methyl Complexes in the Dimerization of Phenylacetylenea
Under the given reaction conditions, PN1 achieved the best conversion (75%) and a Z-selectivity of 95%. A massive drop in reactivity was observed when the cyclohexyl groups were exchanged with isopropyl groups on the P-donor (PN2). Moving from methyl to pyrrolidine substituents on the nitrogen donor led to an additional loss in activity (PN3, PN4, PN7). By replacing the substituents on the nitrogen atom with ethyl groups, the conversion drops to a few precent (PN6). Furthermore, no reactivity of PN5 was detected. This emphasizes the significance of appropriate substituents on both, the phosphorus and nitrogen atoms. Given the lack of any conversion with aryl phosphines, it is evident that a stronger and sterically more demanding σ-donor, such as alkylphosphine, is required to catalyze this given reaction. Thus far, PP3 still performed the best in this transformation, exhibiting excellent conversion and Z-selectivity.
Nonetheless, PN1 outperformed PP2 in terms of conversion and selectivity. Regarding migratory insertion, a propyl group is known to have a greater rate than a methyl group; therefore, a comparison of methylated species would be more equitable.^38,39^ When the methylated compounds are compared, it becomes evident that the PN-supported complex PN1 is superior to PP-supported PP4, emphasizing the benefits of mixed aminophosphine bidentates. Furthermore, no catalytic transformation was observed when utilizing PP5, bearing two triphenylphosphine monodentate ligands. Encouraged by these findings, we decided to screen a series of conditions to improve the performance of PN1. The best result was obtained in EtOH (95%), when compared to MeOH, i-PrOH, THF, toluene, ACN, and DCE. A slight change in selectivity was observed, and no geminal product was detected.
Hydroboration of 4-Chlorostyrene
Next, selected PN complexes were tested for the hydroboration of 4-chlorostyrene. The results are summarized in Table 3. Conducted at a catalyst loading of 1.00 mol %, the hydroboration of 4-chlorstyrene was enabled by all synthesized catalysts with a high selectivity toward the anti-Markovnikov product A. Contrary to the dimerization of phenylacetylene, the use of phenyl groups manifested the best results, reaching an excellent conversion rate of 99% when utilizing PN5. Hence, this complex performed almost as well as the previously reported PP complex PP3.^18^ A similar reactivity was observed for PN6 (93% conversion), followed by PN2 (67% conversion) and PN1 (41% conversion).
Table 3: Catalytic Performance of PN- and PP-Based Mn(I) Methyl Complexes in the Hydroboration of 4-Chlorostyrene
Interestingly, the pyrrolidine-based complexes PN3, PN4, and PN7 performed poorly with a conversion of 22, 16, and 43%, respectively. These findings indicate that the choice of substituents regarding the nitrogen atom is more important than for the phosphorus atom. Good conversion was achieved with complex PP4 (80%) and PP5 (87%). Nonetheless, a suitable combination of mixed PNs, such as in PN5, can outperform mono- and bisphosphine-based Mn(I) alkyl carbonyl complexes, emphasizing, again, the advantage of an unsymmetrical donor set.
Conclusions
In summary, we developed a procedure to synthesize PN Mn(I) alkyl carbonyl complexes. The bromide species were readily transformed into the triflate analogues using AgOTf. MeLi and MeMgCl were used as alkylation agents for synthesizing initial PN-supported Mn(I) methyl carbonyl compounds. The crystal structures of 1b, 1i, 2g, and PN1 verified the octahedral coordination sphere with fac-arranged carbonyls, which is in alignment with the IR data. Preliminary investigations revealed that fac-[Mn(P^Cy^N^Me^)(CO)_3_Me] (PN1) is the best precatalyst for the dimerization of phenylacetylene. In addition, we demonstrated the hydroboration of 4-chlorstyrene, facilitated by all described complexes with a preferred anti-Markovnikov product formation. Especially, fac-[Mn(P^Ph^N^Me^)(CO)_3_Me] (PN5) gave excellent conversion and high selectivity.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1a Hagen J.Industrial Catalysis: A Practical Approach; Wiley, 2005.
- 2Zoeller J. R.; Agreda V. H.; Cook S. L.; Lafferty N. L.; Polichnowski S. W.; Pond D. M. Eastman Chemical Company Acetic Anhydride Process. Catal. Today 1992, 13, 73–91. 10.1016/0920-5861(92)80188-S. · doi ↗
- 3Jahangiri H.; Bennett J.; Mahjoubi P.; Wilson K.; Gu S. A Review of Advanced Catalyst Development for Fischer–Tropsch Synthesis of Hydrocarbons from Biomass Derived Syn-Gas. Catal. Sci. Technol. 2014, 4, 2210–2229. 10.1039/C 4CY 00327 F. · doi ↗
- 4a Ojima I.; Tsai C.; Tzamarioudaki M.; Bonafoux D.The Hydroformylation Reaction. In Organic Reactions; John Wiley & Sons, 2000; pp 1–354.
- 5a Bullock R. M.Catalysis without Precious Metals; Wiley, 2010.
- 6Elangovan S.; Topf C.; Fischer S.; Jiao H.; Spannenberg A.; Baumann W.; Ludwig R.; Junge K.; Beller M. Selective Catalytic Hydrogenations of Nitriles, Ketones, and Aldehydes by Well-Defined Manganese Pincer Complexes. J. Am. Chem. Soc. 2016, 138, 8809–8814. 10.1021/jacs.6b 03709.27219853 · doi ↗ · pubmed ↗
- 7Elangovan S.; Garbe M.; Jiao H.; Spannenberg A.; Junge K.; Beller M. Hydrogenation of Esters to Alcohols Catalyzed by Defined Manganese Pincer Complexes. Angew. Chem., Int. Ed. 2016, 55, 15364–15368. 10.1002/anie.201607233.27690363 · doi ↗ · pubmed ↗
- 8Kallmeier F.; Irrgang T.; Dietel T.; Kempe R. Highly Active and Selective Manganese C = O Bond Hydrogenation Catalysts: The Importance of the Multidentate Ligand, the Ancillary Ligands, and the Oxidation State. Angew. Chem., Int. Ed. 2016, 55, 11806–11809. 10.1002/anie.201606218.27571701 · doi ↗ · pubmed ↗