Morphology, Molecular Characterization, and Phylogeny of Travassosius rufus Khalil, 1922 (Strongylidea: Trichostrongylidae), a Parasite from Endangered Sino-Mongolian Beaver (Castor fiber birulai) in Xinjiang, China
Huiping Jia, Wenwen Chu, Dong Zhang, Kai Li, Wenpu Huang, Xiaoyun Li

TL;DR
This study examines a parasite infecting the endangered Sino-Mongolian beaver, revealing its unique genetic traits and evolutionary history.
Contribution
The first complete mitochondrial genome sequence of the genus Travassosius is reported, providing new insights into its phylogeny.
Findings
The mitochondrial genome of T. rufus is 13,646 bp and contains 36 genes.
Phylogenetic analysis shows T. rufus is closely related to Nematodirus within Trichostrongylinae.
Significant genetic divergence among T. rufus populations is likely due to geographic isolation.
Abstract
The endangered Sino-Mongolian beaver (Castor fiber birulai), a key species for sustaining river ecosystems in China and Mongolia, is threatened by parasitic nematodes. This study focuses on Travassosius rufus, a gastric parasite in these beavers, aiming to understand its characteristics and evolutionary background. For the first time, we sequenced the parasite’s mitochondrial genome and compared samples from China with those from Norway and the Czech Republic. Our findings revealed significant genetic divergence among populations, likely due to prolonged geographic isolation. We also found that while T. rufus shares ancestry with other parasitic worms, it shows features that are uniquely adapted to its beaver hosts. These results contribute to our understanding of how parasites and hosts evolve together. The genus Travassosius Khalil, 1922, the smallest genus in the subfamily…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
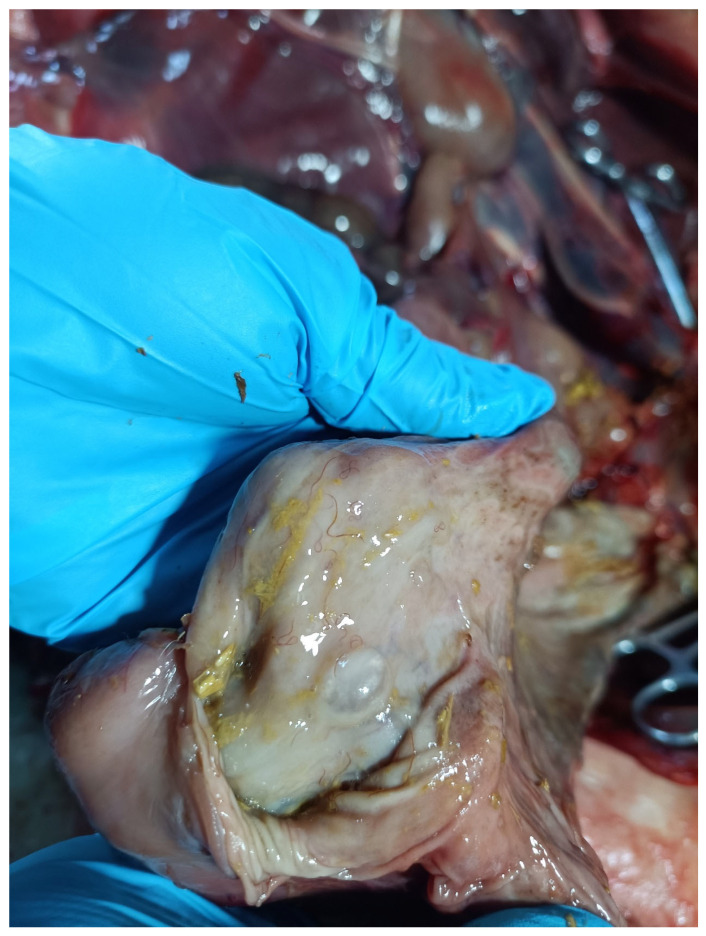 Figure 1
Figure 1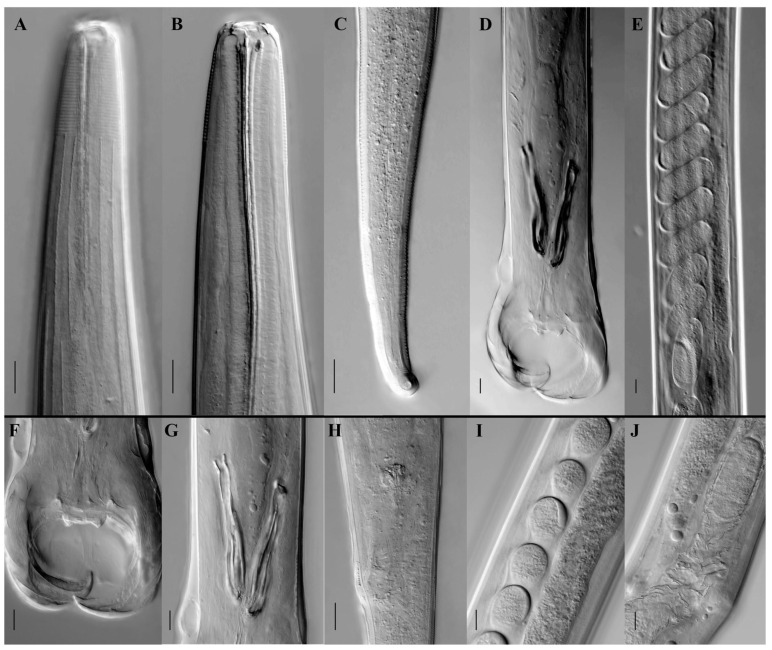 Figure 2
Figure 2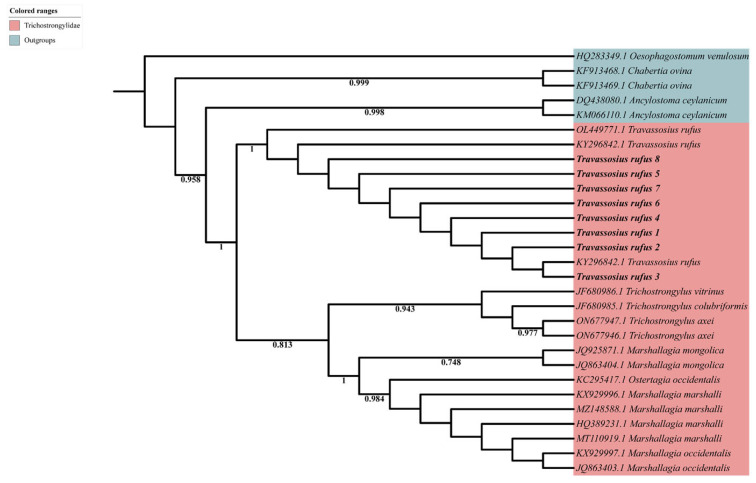 Figure 3
Figure 3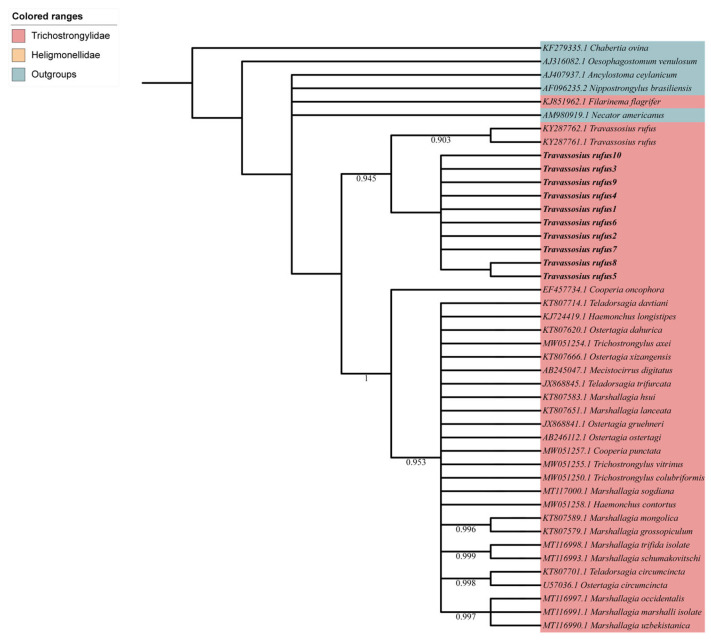 Figure 4
Figure 4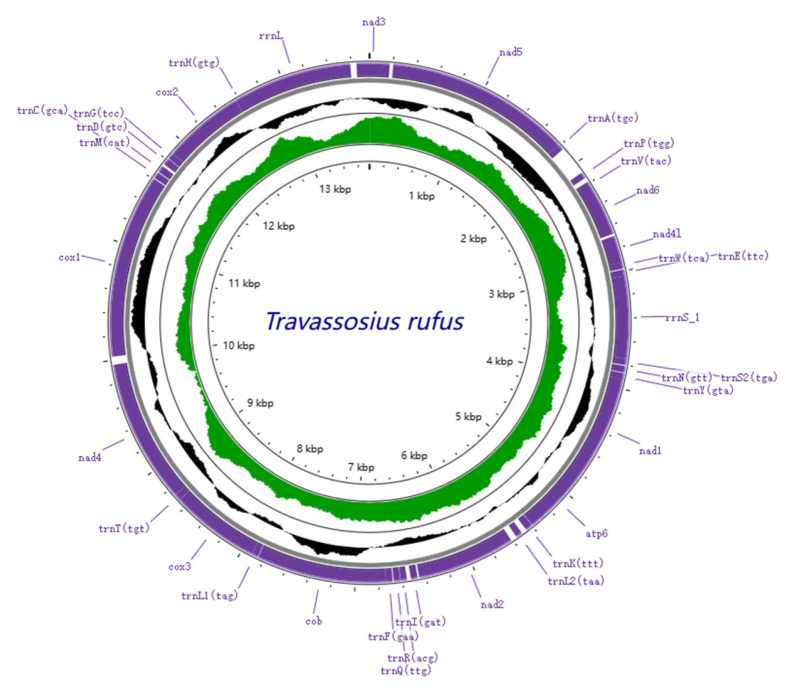 Figure 5
Figure 5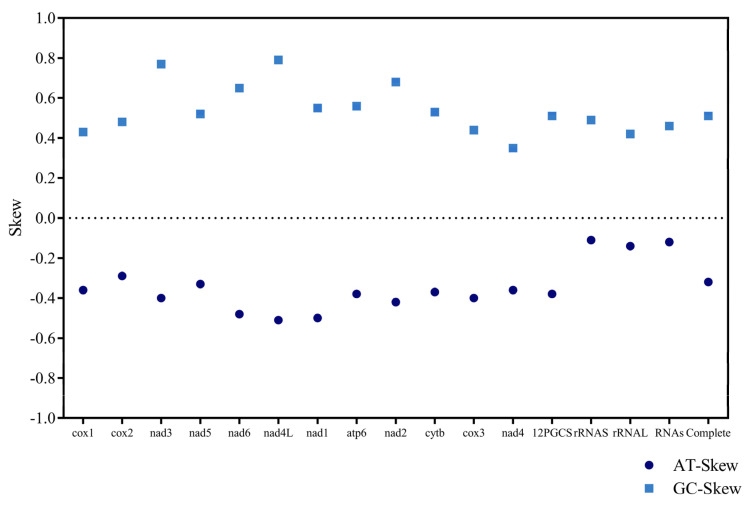 Figure 6
Figure 6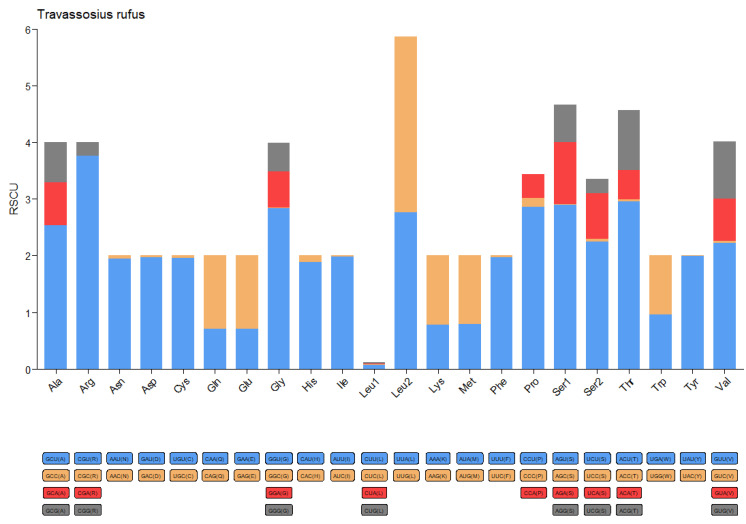 Figure 7
Figure 7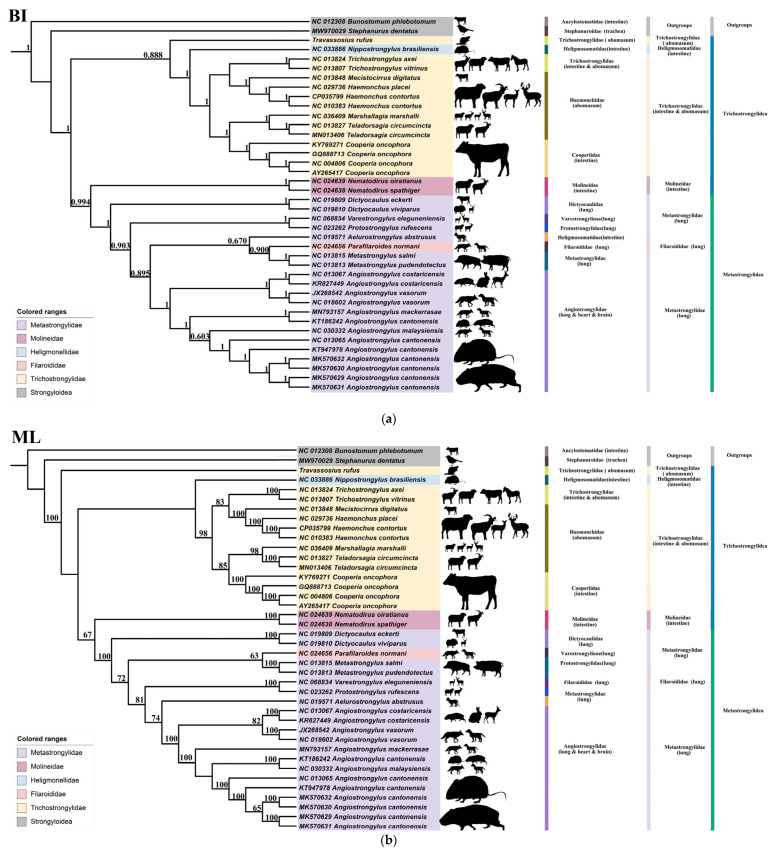 Figure 8
Figure 8- —National Key R&D Program of China
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsEcology and biodiversity studies · Aquatic Invertebrate Ecology and Behavior · Genetic diversity and population structure
1. Introduction
Beavers (Castor spp.) belong to the order Rodentia and the family Castoridae. Globally, the genus Castor includes only two living species: the North American beaver (C. canadensis), which is widespread across North America, and the Eurasian beaver (C. fiber) [1,2], which once inhabited most river systems throughout northern Eurasia [3]. The Eurasian beaver serves a key ecological function in freshwater systems across Europe and Asia. Its dam-building behavior not only controls water flow [4] but also forms wetland habitats [5], which support greater biodiversity [6,7,8]. The Sino-Mongolian beaver, a subspecies of the Eurasian beaver, is native to the Ulungur River Basin in northern Xinjiang, China, and the adjoining upstream regions along the Mongolian border. It is listed as a first-class nationally protected wild animal in both countries. As a keystone species in the oasis-valley mosaic ecosystem characteristic of arid regions, the Sino-Mongolian beaver plays an essential role in maintaining ecological stability in the region [1,9].
Historically, overhunting of Eurasian beavers for fur and castoreum led to a dramatic population collapse [10]. While conservation efforts have supported the species’ recovery in some areas, health-related risks continue to constrain stable population growth. Among these threats, parasitic infections have emerged as a major concern for Eurasian beaver conservation [11,12]. Parasitic nematodes, which commonly reside in the gastrointestinal tract of mammals, are prominent pathogens. By absorbing nutrients, they may induce malnutrition, anemia, and sometimes severe systemic conditions [13,14].
Travassosius rufus Khalil, 1922, a nematode species parasitizing the stomach of the Eurasian beaver, belongs to the family Trichostrongylidae and is the type species of the genus Travassosius. In China, this species was first reported by Zhang et al. in 1992 [15]. Subsequent studies have focused mainly on morphological identification. Prior work has shown that nuclear and mitochondrial sequence data, including complete mitochondrial genomes, are essential in the integrative taxonomy, population genetics, and phylogenetic research of Trichostrongylidae nematodes. In recent years, developments in molecular biology have introduced new methods for nematode taxonomy. At present, the NCBI database includes only two mitochondrial COI gene sequences and two nuclear ITS sequences for T. rufus, and there are no records of mitochondrial genomes for the genus Travassosius. To define the full mitochondrial genome features of Travassosius, we sequenced the ribosomal DNA of T. rufus obtained from the Sino-Mongolian beaver, including the mitochondrial COI gene and nuclear ITS2 region, and conducted the first annotation of its mitochondrial genome. To evaluate the taxonomic validity of the genus Travassosius and explore its phylogenetic placement within Trichostrongylidae, we performed phylogenetic analyses based on concatenated amino acid sequences of 12 mitochondrial protein-coding genes (PCGs), using both Maximum Likelihood (ML) and Bayesian Inference (BI) approaches. This study offers a scientific foundation for conserving the endangered Sino-Mongolian beaver in China and Mongolia and for addressing parasitic infections within its population.
2. Materials and Methods
2.1. Sample Collection
In May 2024, a Sino-Mongolian beaver rescued from the wild due to malnutrition and extreme emaciation died at the Fuyun County Terrestrial Wildlife Epidemic Prevention and Rescue Station (46.99419326° N, 89.55346159° E). A necropsy was conducted with the assistance of professional veterinarians, with particular attention given to examining the digestive tract for parasitic infections. Nematode specimens collected from the stomach were rinsed in physiological saline (0.9% NaCl) and preserved in 75% ethanol for morphological and molecular analyses. All recovered nematodes were identified as belonging to a single species. Samples were stored at −40 °C for DNA extraction and subsequent analyses.
2.2. Morphological Observation
Specimens were cleared with lactophenol to improve the visibility of internal structures. The prepared samples were mounted on glass slides and covered with coverslips. The morphological traits of adult worms were observed and documented under a Carl Zeiss differential interference contrast microscope (Axioscope 5, Baden-Württemberg, Germany) at 10× and 40× magnifications. The recorded characteristics included body length, body width, esophagus length, and sex-specific traits such as male and female tail morphology. All measurements were expressed in micrometers (μm), and statistical analyses were conducted to assess inter-individual variability. We then examined the adult nematodes under a microscope and recorded the number of nematodes.
2.3. Molecular Analysis
2.3.1. DNA Extraction, PCR Amplification, and Sequencing
Adult nematode samples stored at −40 °C were used for total genomic DNA extraction using the DNeasy Blood and Tissue Kit protocol. Specific primers targeting multiple genetic markers (18S, ITS, cox1) were designed (Table 1), and PCR amplification was carried out to obtain the corresponding fragments. PCR products were verified using 2% agarose gel electrophoresis and sent to BGI (Beijing Genomics Institute, Beijing, China) for Sanger sequencing. In addition, whole-genome sequencing was performed on the Illumina NovaSeq 6000 PE150 platform using remaining amplified DNA. Sanger sequencing results were employed as bait sequences, and the resulting data were used for genome annotation and bioinformatic analysis.
2.3.2. DNA Annotation and Data Analysis
Sequence Analysis
Gene sequences obtained via Sanger sequencing from BGI were compared to the NCBI database using BLAST (Basic Local Alignment Search Tool, v 2.13.0) to identify homologous sequences. Relevant sequences from closely related species within the same genus were downloaded for downstream analysis. Multiple sequence alignments were conducted using the ClustalW algorithm implemented in MEGA 7.0 under default settings. Manual inspection was carried out to exclude incomplete or low-quality terminal regions, ensuring reliable alignments. To reduce background noise and retain informative segments, alignments were trimmed to preserve conserved regions with high-quality signals. Pairwise genetic distance matrices were generated for the 18S rRNA sequences to assess intraspecific variability.
Following sequence alignment and trimming, phylogenetic analysis was performed using the Maximum Likelihood (ML) method on ITS and COI sequences. The Tamura-Nei (TN93) model was selected as the best-fit substitution model. Node support was evaluated through bootstrap analysis with 1000 replicates. The resulting phylogenetic tree was visualized and edited using ITOL (https://itol.embl.de/, accessed on 26 March 2025).
Sequencing data were subjected to quality control, de novo assembly, and annotation via the IDBA assembler. Bait sequences were applied to extract the mitochondrial genome. Partial mitochondrial genome data from other Trichostrongylidae species were obtained from NCBI for comparative purposes. The annotated mitochondrial genome of Travassosius rufus was visualized using the Proksee Server (https://proksee.ca/, accessed on 10 December 2024). Further mitochondrial genome analysis included the assessment of nucleotide composition and codon usage across the 12 protein-coding genes (PCGs), excluding stop codons. Relative synonymous codon usage (RSCU) was calculated following the approach of Tamura et al. (2011) [19] using MEGA. AT and GC skews were computed based on the method by Perna and Kocher (1995) [20]. Graphs and figures were produced using the ggplot2 package (v3.4.1) in R and visualized using Prism GraphPad (Prism 9, Boston, MA, USA).
2.4. Phylogenetic Tree Construction
Phylogenetic analysis was carried out for both Travassosius species and other Trichostrongylidae nematodes to clarify the evolutionary placement and taxonomic distinctiveness of T. rufus. Twelve protein-coding gene (PCG) fragments were selected for this purpose. Species from the Cooperiidae family, including Cooperia oncophora, along with members of the Habronematidae family, were used as outgroups (Supplementary Table S1, [21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43]). Maximum Likelihood (ML) analysis was performed using RAxML version 8, while Bayesian Inference (BI) analysis was carried out with MrBayes version 3.2.7a, [44]. The resulting phylogenetic trees were used to investigate systematic relationships within Trichostrongylidae. Individual PCG fragments were aligned with MAFFT (version 7.037), concatenated using SequenceMatrix (version 1.7.8), and visualized on the iTOL online platform (https://itol.embl.de/, accessed on 26 March 2025). In addition to tree construction, host–parasite associations were examined. Host data were compiled, including the relationship between T. rufus and Sino-Mongolian beaver observed in this study, along with previously reported host records for Trichostrongylidae species from public databases. The taxonomic positions of parasitic species were defined using both ML and BI trees, and host–parasite interactions were mapped accordingly to illustrate these relationships more clearly.
3. Results
3.1. Morphological and Taxonomic Study of Travassosius rufus in Sino-Mongolian Beaver Based on Classification Methods
A total of 5478 adult nematodes were recovered from the stomach of the Sino-Mongolian beaver during dissection (Figure 1). Morphological identification confirmed the species as T. rufus, distinguished by its small, slender form and brick-red coloration. The body is cylindrical, with the greatest width located just behind the midpoint. The anterior and posterior ends of the cuticle show distinct transverse striations, while the rest of the body surface bears longitudinal ridges (Figure 2A). The buccal capsule is small (Figure 2B), and the cervical papillae are prominent. The pre-bursal papillae are large and well-defined, with a broad base and rounded apex. In males, the lateral bursal rays are elongated, while the dorsal rays are comparatively short. A pair of spicules is present, but there is no gubernaculum. Female specimens exhibit a tail that gradually narrows and curves dorsally at the end (Figure 2C). The distance from the tip of the tail to the vulva is shorter than the distance from the vulva to the anus.
Male specimens (based on measurements and observations from three mature individuals; Table 2, Figure 2): Body length ranges from 8.69 to 10.477 mm (mean: 9.68 mm), with a maximum body width of 0.112–0.184 mm (mean: 0.140 mm). The buccal capsule is simple. The cuticle presents 31–32 transverse striations and 30–32 longitudinal ridges. The esophagus measures 0.452–0.552 mm (mean: 0.510 mm), accounting for 4.31–6.35% (mean: 5.27%) of total body length. The nerve ring is situated 0.272–0.340 mm (mean: 0.292 mm) from the anterior end, and the excretory pore lies 0.310–0.385 mm (mean: 0.330 mm) from the anterior end. Cervical papillae are positioned 0.396–0.479 mm (mean: 0.428 mm) from the anterior tip. The bursa margin is undulated, with relatively large lateral lobes and smaller dorsal lobes that are not clearly separated. Lateral lobes typically curve ventrally, forming an embracing pattern. Pre-bursal papillae are large and distinct, measuring approximately 0.015 mm in length, with a broad base and a rounded apex. The paired spicules (Figure 2D,G) are thick and split into two branches near the mid-region. The lateral branch runs closely alongside the median branch, with their distal ends nearly touching. This feature sets T. rufus apart from its congener T. americonus, where the lateral and median branches of the spicules are more widely spaced. A gubernaculum is absent. The copulatory cone is stout and nearly conical, with numerous bubble-like projections on its surface.
Female specimens (based on measurements and descriptions of three mature individuals; Table 2, Figure 2): Body length ranges from 9.89 to 11.87 mm (mean: 11.24 mm), with maximum body width between 0.142 and 0.161 mm (mean: 0.157 mm). The cuticle shows 31–32 transverse striations and 32–36 longitudinal ridges. The esophagus measures 0.512–0.559 mm (mean: 0.544 mm), accounting for 4.31–5.65% (mean: 4.84%) of total body length. The nerve ring is located 0.260–0.282 mm (mean: 0.271 mm) from the anterior end, and the excretory pore lies 0.332–0.396 mm (mean: 0.358 mm) from the anterior tip. Cervical papillae are situated 0.436–0.468 mm (mean: 0.454 mm) from the anterior end. The vulva is positioned 2.247–2.831 mm (mean: 2.649 mm) from the tail tip (Figure 2H). The tail is long and thin and curves dorsally at the posterior end, terminating in a blunt tip. Transverse striations are visible along the tail surface. Eggs are elongated-oval in shape (Figure 2E,I), with a thin, smooth shell, measuring 78.10–89.56 μm (mean: 81.89 μm) in length and 40.18–52.78 μm (mean: 44.10 μm) in width (Table 2, Figure 2).
3.2. Molecular Taxonomy and Phylogenetic Position of Travassosius rufus in Sino-Mongolian Beaver Based on Sanger Sequencing
The genetic variation of nematodes infecting the Sino-Mongolian beaver was analyzed using the 18S rRNA gene. All 18S sequences showed high similarity, with a length of 1577–1578 bp, indicating strong sequence conservation, consistent with the gene’s role as a core ribosomal component. A genetic distance matrix revealed very low divergence among most samples (0.000–0.003), particularly between samples 2, 5, and 6, where the distance was 0.000 (Supplementary Table S2), suggesting a genetically homogeneous or clonal origin. However, sample 1 showed a significantly greater genetic distance (0.016–0.018), exceeding the typical intraspecific variation threshold (>1%). This could suggest potential taxonomic divergence (e.g., a cryptic species) or technical artifacts such as sequence contamination.
Sanger sequencing revealed that the ITS2 gene sequence of T. rufus samples was 419–455 bp in length. Alignment with T. rufus sequences available in the GenBank database showed consistently high similarity, exceeding 97.97%. Similarity with other sequences was below 90%. A phylogenetic tree based on nuclear ITS2 gene sequences (Figure 3) was constructed to examine the genetic relationships among T. rufus samples from various geographic locations. Tree branches were color-coded by family for visualization. Travassosius rufus samples (T. rufus 1–8) from the Sino-Mongolian beaver in China clustered with previously published sequences from C. fiber in Norway (KY296842.1, KY287762.1) and the Czech Republic (OL449771.1), with strong support (bootstrap = 1), indicating high sequence similarity across regions. Phylogenetic analysis placed T. rufus in a distinct and well-supported clade within Trichostrongylidae, as sister to Trichostrongylus (support = 0.813), and clearly separated from Marshallagia and Ostertagia. No evident clustering by host or geography was observed among T. rufus samples, indicating low intraspecific variation in the ITS2 region and supporting its status as a valid, phylogenetically distinct species.
The phylogenetic tree based on mitochondrial COI sequences (Figure 4) was constructed to explore the evolutionary relationships among T. rufus samples and related nematode species. Consistent with the ITS2-based findings, multiple T. rufus sequences clustered into a strongly supported monophyletic branch (support value = 0.903), indicating molecular coherence and evolutionary separation at the species level. T. rufus samples from the Sino-Mongolian beaver in China grouped into a clade alongside sequences from Norway (KY287761.1, KY287762.1), suggesting substantial genetic similarity among geographically distant populations. This branch has a clear differentiation boundary from other genera and species in the Trichostrongylidae family (indicated by the pink background), such as Trichostrongylus, Ostertagia, and Marshallagia. In the phylogenetic tree, the branch where Travassosius rufus did not form a sister group relationship with any specific genus but exists as an independent evolutionary lineage within the Trichostrongylidae family suggests that its phylogenetic position is relatively independent and is further supported at the molecular level based on morphological classification. The outgroups Ancylostoma ceylanicum, Necator americanus, Oesophagostomum venulosum, Chabertia ovina, and Nippostrongylus brasiliensis successfully anchored the root of the tree, supporting the overall phylogenetic structure and evolutionary relationships among the species analyzed.
3.3. Phylogenetic and Micro-Taxonomic Classification of Travassosius rufus Based on Whole-Genome Sequencing
The complete mitochondrial genome of Travassosius rufus was sequenced, revealing a total length of 13,755 bp. It includes 12 protein-coding genes (cox1–3, nad1–6, nad4L, atp6, atp8, and cytb), 22 tRNA genes, and 2 rRNA genes (Table 3, Figure 5). The mitochondrial genome size of T. rufus is comparable to those of other Trichostrongylidae species, such as Trichostrongylus colubriformis (13,653 bp) and Trichostrongylus vitrinus (13,800 bp). All genes are encoded on the H-strand (positive strand) (Table 4, Figure 5). The mitochondrial DNA of T. rufus shows a strong AT bias (74.0%), with thymine (T) being the most frequent nucleotide (51.1%) and cytosine (C) the least (6.0%). The overall AT-skew and CG-skew are −0.314 and 0.510, respectively. The combined length of the 12 protein-coding genes is 10,296 bp, making up 75.9% of the total mitochondrial genome (Table 4). These genes also show a high AT content, ranging from 67.5% to 77.2%, with nad6, nad4, and nad5 showing the highest A+T content at 76.4%, 76.7%, and 77.2%, respectively (Table 4). The AT-skew values for nad6, nad4, and nad5 are −0.48, −0.36, and −0.33, and the GC-skew values are 0.65, 0.35, and 0.52, respectively (Table 4, Figure 6). Together, the 12 protein-coding genes encode 3429 amino acids (excluding stop codons). Codon usage analysis identified CGU (Arg), UUG (Leu), and ACU (Thr) as the most frequently used codons in the T. rufus mitochondrial genome (Figure 7). Additionally, 22 typical tRNA genes were annotated, ranging from 55 to 64 bp in length, all encoded on the H-strand. The mitochondrial genome also contains two rRNA genes: rRNAS (703 bp) and rrnL (982 bp). The rrnS gene is situated between tRNA-Glu and tRNA-Ser2, while rrnL lies between tRNA-His and the protein-coding gene nad3. The A+T content of rrnS and rrnL is 74.8% and 77.8%, respectively, with an overall RNA A+T content of 76.3%.
This study constructed BI and ML phylogenetic trees based on 12 mitochondrial PCGs. The tree topologies produced by both methods were highly consistent, clearly resolving phylogenetic relationships across families such as Trichostrongylidae, Metastrongylidae, and Haemonchidae, and forming well-supported monophyletic clades. In the BI tree, T. rufus was positioned within Trichostrongylidae and grouped as a sister taxon to Nipostrongylus brasiliensis, with a support value of 0.888. This suggests that T. rufus may share a recent common ancestor with N. brasiliensis or exhibit similar host-related evolutionary traits. The placement of T. rufus aligned with other Trichostrongylidae members, reinforcing its classification within this family (Figure 8a). In the ML tree, T. rufus also appeared within Trichostrongylidae and again formed a sister group with N. brasiliensis (Figure 8b), confirming the consistency of the BI results. These outcomes demonstrate that the assignment of T. rufus to Trichostrongylidae is stable and its evolutionary relationships are consistently supported across different analytical methods. Compared to other species within Trichostrongylidae, T. rufus occupies a separate evolutionary branch, indicating it may have undergone distinct evolutionary changes linked to host adaptation.
4. Discussion
4.1. Taxonomy of Travassosius rufus in Beavers Based on Morphology
The genus Travassosius comprises two recognized species: T. rufus Khalil, 1922, and T. americanus Chapin, 1925 [45], which parasitize the stomachs of Eurasian and North American beavers, respectively. However, the classification of these two species has long been debated. Cameron (1938) regarded T. americanus as a junior synonym of T. rufus, arguing that both names refer to the same species [46]. In contrast, researchers such as Smith (1967) maintained that T. rufus and T. americanus should be treated as distinct species [47]). In 1978, Bush and Samuel analyzed specimens collected from Alberta, Pennsylvania, British Columbia, and Europe [48]. By comparing the type specimens of T. americanus and T. rufus, they identified pronounced morphological differences between parasites infecting North American and Eurasian beavers. In particular, they noted distinct differences in the length and structure of the male spicules, supporting the recognition of T. americanus and T. rufus as separate species. They further proposed that T. americanus, which infects North American beavers, represents the ancestral lineage, while T. rufus, found in Eurasian beavers, emerged through geographic isolation. Their work also raised the possibility that the T. americanus population on Vancouver Island might be undergoing speciation [48]. Based on the morphological traits described by Zhang Meiyun et al. and Bush et al., we identified the nematodes collected from the Sino-Mongolian beaver in this study as T. rufus [15,48]. Further confirmation was achieved using differential interference contrast (DIC) microscopy, which allowed for clearer observation of diagnostic features such as transverse rings on the body surface, longitudinal striations, spicules, and egg morphology. This approach enabled a more detailed evaluation of key taxonomic characteristics, reinforcing the identification of T. rufus.
4.2. Molecular Taxonomy and Phylogenetic Status of Travassosius rufus Based on DNA Barcoding
Based on the research results, this study revealed the genetic characteristics of T. rufus and its taxonomic status within the family Trichostrongylidae from a molecular phylogenetic perspective. Phylogenetic analyses based on the nuclear ITS2 and mitochondrial COI sequences both demonstrated that T. rufus formed a strongly supported monophyletic clade within Trichostrongylidae, with clear evolutionary boundaries separating it from closely related genera such as Trichostrongylus, Ostertagia, and Marshallagia. These molecular findings corroborate the validity of T. rufus as a distinct species, originally established through morphological characteristics. Notably, despite differences in evolutionary rates between ITS2 and COI markers, both genes revealed high intraspecific sequence conservation in T. rufus: ITS2 sequences exhibited >97.97% similarity across geographically distributed populations, while COI clustering indicated low genetic divergence among populations from China, Norway, and the Czech Republic. This pattern suggests that T. rufus may possess strong host adaptability, with its dispersal potentially linked to host migration history and niche conservatism.
In phylogenetic reconstructions, T. rufus did not form a sister-group relationship with any specific genus but instead occupied an independent evolutionary lineage within Trichostrongylidae, possibly reflecting its unique evolutionary trajectory or the existence of undisclosed closely related taxa. The outgroup species (e.g., Oesophagostomum venulosum and Chabertia ovina) successfully anchored the root of the phylogenetic tree, enhancing the reliability of the topological structure, though caution is warranted regarding the limitations of mitochondrial genes in resolving deep-level phylogenetic relationships. Overall, the genetic data obtained for T. rufus in this study will serve as a critical reference for future research on species identification, population genetics, and phylogenetic investigations of this taxon.
4.3. Molecular Taxonomy and Phylogenetic Status of Travassosius rufus Based on Genome Sequencing
This study presents the first complete mitochondrial DNA (mtDNA) genome of T. rufus parasitizing the Sino-Mongolian beaver in China. Comparative analysis with other Trichostrongylidae species, such as Trichostrongylus colubriformis (13,693 bp) and Trichostrongylus axei (13,653 bp), showed that the mtDNA genome length of T. rufus is similar, indicating a degree of size conservation across the family. Nucleotide composition analysis exhibited a strong AT bias (73.9%) in the T. rufus mtDNA genome, especially within PCGs, where the A+T content reached 75.9% (Table 4). Notably, nad5 (77.2% AT) and nad6 (76.4% AT) exhibited the highest AT content, with negative AT-skew (−0.33 and −0.48, respectively) and positive GC-skew (0.52 and 0.65), reflecting strand-specific mutational pressures from unidirectional replication and limited DNA repair [49,50,51]. This compositional bias aligns with observations in other nematodes [52,53] and may facilitate metabolic adaptation to parasitic niches by reducing transcriptional energy costs [54] and tolerating mutation accumulation under host oxidative stress. Similar to other members of Trichostrongyloidea, including Mecistocirrus digitatus (15,221 bp) [21] and Haemonchus contortus [55], the T. rufus mitochondrial genome lacks the atp8 gene. All 12 PCGs in T. rufus are encoded on the heavy strand and transcribed in the same direction, consistent with findings in other Trichostrongylidae species (see Figure 8). The PCGs of T. rufus mainly use standard start codons such as ATT and TTG, with common termination codons including TAA and TAG. Some genes show alternative patterns: cox2, nad3, cytb, and nad4 begin with GTT, ATG, ATA, and GTG, respectively. In several cases, genes like nad5 end with incomplete stop codons (e.g., T). These incomplete stop codons are typical in nematode mitochondrial genomes [25,26].
In the mitochondrial DNA genome of Travassosius rufus, certain codons—such as CGU (Arg), UUG (Leu), and ACU (Thr)—show a clear usage preference. This pattern is consistent with findings from studies on the Haemonchus genus and other nematodes [26,28]. The codon usage bias (e.g., CGU/UUG dominance) and high AT content in rRNA genes (76.3% overall) further align with energy-efficient genome streamlining, balancing rapid adaptation and functional conservation [24,56]. This supports the research of Gendron et al. (2024) [57], where niche-specific pressures (e.g., parasitism) and phylogenetic constraints jointly shape nematode mitogenome plasticity [51,54]. Codon preference analysis in this study highlights differences in gene expression control and protein synthesis between T. rufus and other nematode species. These distinctions suggest that T. rufus, as a strict parasite of the Sino-Mongolian beaver, may have developed unique adaptive traits over its evolutionary history, allowing it to remain well-suited to varying host environments. Both Bayesian Inference (BI) and Maximum Likelihood (ML) analyses in this study place T. rufus within the family Trichostrongylidae, forming a sister group with Nipostrongylus brasiliensis. This indicates the possibility of a shared common ancestor. This taxonomic position is in line with earlier morphological classifications, further supporting the stable placement of T. rufus within Trichostrongylidae. The evolutionary trajectory of T. rufus may have been shaped by host specificity or environmental selection. Further investigation is needed to better understand its host adaptation and ecological dynamics.
The host specificity of T. rufus and its possible evolutionary route are particularly noteworthy. T. rufus primarily infects beavers (Castoridae), which contrasts with the usual host range of most Trichostrongylidae species. For example, N. brasiliensis parasitizes rodents, while many other Trichostrongylidae species infect ruminants or equids. In both the BI and ML phylogenetic trees, T. rufus appears closely related to N. brasiliensis, suggesting that it may represent a specialized lineage adapted to rodent hosts. The adaptation of T. rufus to beavers could be the result of a host-switching event, or it is possible that its ancestors infected a broader range of hosts before gradually specializing in beavers. This study presents new molecular evidence for the systematics of T. rufus, based on its mitochondrial genome and the full set of 12 protein-coding genes. Mitochondrial genomes, due to their relatively high mutation rates and maternal inheritance, provide strong resolution in phylogenetic studies of parasitic nematodes. The agreement between BI and ML results supports the reliability of mitochondrial data for resolving the phylogenetic position of T. rufus. Future work should incorporate nuclear gene markers (e.g., ITS, 18S, and 28S rRNA) to refine the evolutionary framework and clarify the lineage history of T. rufus.
5. Conclusions
This study investigates the structural and genomic features of Travassosius rufus, a parasitic nematode exclusive to the endangered Sino-Mongolian beaver, through morphological, genetic, and phylogenetic analyses. It fills a key gap in mitochondrial genome data for T. rufus. Phylogenetic trees constructed using ML and BI approaches confirm that T. rufus from the Sino-Mongolian beaver shows significant genetic separation from other species. Its evolutionary history suggests a pattern of co-evolution with its host. DNA markers (e.g., ITS2, cox1) identified here offer practical tools for non-invasive monitoring of parasite prevalence and genetic diversity, critical for detecting infection risks during population recovery efforts. Given the strong host specificity of T. rufus, uncontrolled parasite loads may exacerbate conservation challenges for this keystone species. Integrating genomic insights into management strategies—such as routine parasite screening in introduction programs—could mitigate declines. Future studies should prioritize quantifying the link between T. rufus infection intensity and host health while exploring parasite adaptation mechanisms to refine conservation measures. These findings underscore the importance of parasite surveillance and investigation in wildlife, particularly for ecologically pivotal hosts like the Sino-Mongolian beaver.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Lu H. The distribution and ecological characteristics of the beaver (Castor fiber L.) in China J. Shandong Univ. (Nat. Sci.)19814103109
- 2Chu H. Jiang Z. Distribution and Conservation of the Sino-Mongolian Beaver (Castor fiber birulai) in China Oryx 20094319720210.1017/S 0030605308002056 · doi ↗
- 3Djoshkin W. Safonow W. Die Biber der Alten und Neuen Welt Ziemsen Verlag Wittenberg, Germany 1972
- 4RaškauskaitėM. Šimkevičius K. Eurasian Beaver (Castor fiber L.) Population in Asu Science and Teaching Hunting Area and Beaver Dams Rebuild Intensity Vytautas Magnus University Kaunas, Lithuania 2018
- 5Gridan A. NițăD. Ionescu G. Popa M. Pașca C. Distribution, structural and functional characteristics of beaver dams (Castor fiber): Case study Black River Basin 2017228892
- 6Nica A. Petrea M.-S. Simionov I.-A. Antache A. Cristea V. Ecological impact of european beaver Castor fiber. Anim. Sci.202265640647
- 7Brazier R.E. Puttock A. Graham H.A. Auster R.E. Davies K.H. Brown C.M.L. Beaver: Nature’s Ecosystem Engineers Wires Water 20218 e 149410.1002/wat 2.149433614026 PMC 7883483 · doi ↗ · pubmed ↗
- 8Bashinskiy I.V. Beavers in Lakes: A Review of Their Ecosystem Impact Aquat. Ecol.2020541097112010.1007/s 10452-020-09796-4 · doi ↗