Late Presentation of Galloway-Mowat Syndrome (GAMOS) Associated With Membranous Nephropathy: A Case Report
Eugene K Yeboah, Steven Salvatore, Sandeep Sasidharan, Sulayman Khan, Subodh Saggi

TL;DR
An elderly man was diagnosed with a rare genetic disorder, Galloway-Mowat syndrome, linked to kidney disease and unusual proteinuria patterns.
Contribution
This is the first reported case of GAMOS in an adult presenting with PLA2R-negative membranous nephropathy.
Findings
The patient had a WDR73 gene deletion and membranous nephropathy with atypical features.
Immunofluorescence showed IgG and C3 deposits but no PLA2R or other common markers.
Electron microscopy revealed subepithelial and intramembranous electron-dense deposits.
Abstract
We present a rare case of Galloway-Mowat syndrome (GAMOS) in an elderly patient with a WD repeat domain 73 (WDR73) gene deletion. A 64-year-old man with recurrent deep vein thrombosis on anticoagulation, intermittent atrial fibrillation, ulcerative colitis, apical hypertrophic cardiomyopathy, hypertension, and biopsy-proven membranous nephropathy (MN) presented with urinary frequency associated with frothing. His physical examination was unremarkable. His workup revealed worsening proteinuria, which was previously controlled with tacrolimus, low-dose steroid, and enalapril. His laboratory workup showed a serum creatinine level of 1 mg/dL, albumin at 3.5 g/dL, a urine-protein-to-creatinine ratio of 2,800 mg/g, complement 3 (C3) at 127 mg/dL, and complement 4 at 41 mg/dL. Although APOL1 testing was negative, a pathologic deletion in the WDR73 gene was identified. Repeat kidney biopsy…
Genes, proteins, chemicals, diseases, species, mutations and cell lines named across the full text — each resolved to its canonical identifier and authoritative record.
Click any figure to enlarge with its caption.
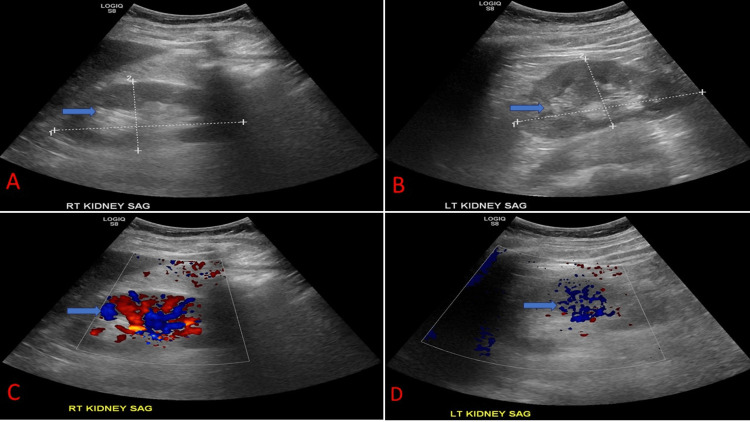 Figure 1
Figure 1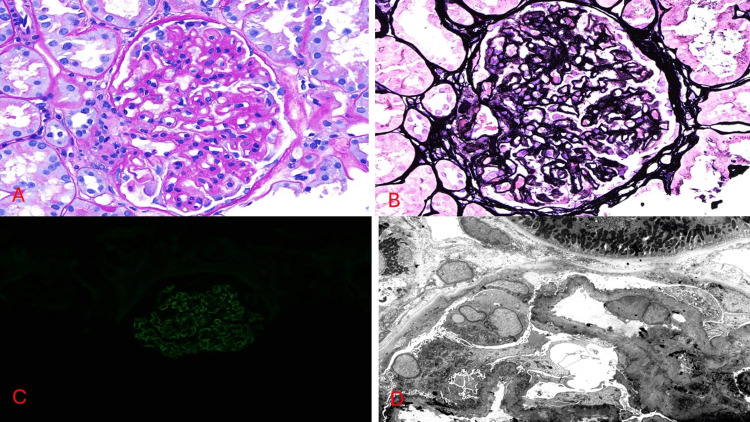 Figure 2
Figure 2| Parameter | Patient values | Reference range |
| Comprehensive metabolic panel | ||
| Sodium | 138 mmol/L | 136-145 mmol/L |
| Potassium | 3.9 mmol/L | 3.5-5.1 mmol/L |
| Calcium | 8.7 mg/dL | 8.2-10 mg/dL |
| Chloride | 103 mmol/L | 98-107 mmol/L |
| Magnesium | 1.6 mg/dL | 1.9-2.7 mg/dL |
| Creatinine | 1.0 mg/dL | 0.7-1.3 mg/dL |
| Blood urea nitrogen | 12 mg/dL | 7-25 mg/dL |
| Carbon dioxide | 28 mmol/L | 21-31 mmol/L |
| Glucose | 93 mg/dL | 70-99 mg/dL |
| Anion gap | 11 mmol/L | 10-20 mmol/L |
| Estimated glomerular filtration rate | 82 mL/minute/1.73 m² | >60 mL/minute/1.73 m² |
| Liver function test | ||
| Total bilirubin | 0.6 mg/dL | 0.3-1 mg/dL |
| Albumin | 3.5 g/dL | 3.5-5.7 g/dL |
| Total protein | 5.8 g/dL | 6-8.3 g/dL |
| Aspartate aminotransferase | 21 U/L | 13-39 U/L |
| Alanine aminotransferase | 13 U/L | 7-52 U/L |
| Alkaline phosphatase | 82 U/L | 34-104 U/L |
| Glycated hemoglobin (HbA1C) | 4.6% | <5.7% |
| Complete blood count | ||
| Hemoglobin | 14.6 g/dL | 14-18 g/dL |
| White blood count | 8.41k/μL | 3.5-10.8k/μL |
| Platelet | 152k/μL | 130-400k/μL |
| Hematocrit | 43.2% | 42%-52% |
| Urinalysis | ||
| Appearance | Slightly cloudy | Clear |
| pH | 5 | 5-8 |
| Specific gravity | 1.024 | 1.005-1.030 |
| Urine glucose | Negative | Negative |
| Urine blood | Small | Negative |
| Urine creatinine | 282 mg/dL | 20-320 mg/dL |
| Urine protein | >500 mg/dL | Negative |
| Urine nitrite | Negative | Negative |
| Leucocyte esterase | Negative | Negative |
| White blood cells (urine) | <1/hpf | 0-5/hpf |
| Urine cast | 21/lpf | 0-2/lpf |
| Urine protein-creatinine ratio | 2,800 mg/g | <150 mg/g |
| Coagulation | ||
| Prothrombin time | 12.9 seconds | 10.8-13.7 seconds |
| Activated partial thromboplastin time | 31.3 seconds | 25-35 seconds |
| International normalized ratio | 1.1 | <1 |
| Glomerulopathy workup | ||
| Complement (C3) levels | 127 mg/dL | 86-184 mg/dL |
| Complement (C4) levels | 41 mg/dL | 20-58 mg/dL |
| Complement total (CH50) | 57 | 42-95 U/mL |
| Infectious workup | ||
| Human immunodeficiency virus 1/2 antigen/antibodies | Negative | Negative |
| Hepatitis C | Nonreactive | Nonreactive |
| Hepatitis B surface antigen | Nonreactive | Nonreactive |
| Tuberculosis QuantiFERON gold | Negative | Negative |
| Autoimmune workup | ||
| Antinuclear antibody | Negative | Negative |
| Anti-double-stranded DNA | <1 IU/mL | <29 IU/mL |
| Genetic test | ||
| Apolipoprotein L1 | Negative | Negative |
| WDR73 gene deletion | Positive | Negative |
Peer Reviews
No public reviews on file for this paper yet. If you reviewed it on a platform where reviews are public (OpenReview, ICLR, NeurIPS, ICML), you can paste yours below so the community can read it here.
Videos
No videos yet. Explain this paper in a talk, walkthrough, or lecture? Add one.
Taxonomy
TopicsRenal Diseases and Glomerulopathies · Ion Transport and Channel Regulation · Dialysis and Renal Disease Management
Introduction
Galloway-Mowat syndrome (GAMOS) is an uncommon autosomal recessive disorder characterized by various neurological and renal abnormalities [1]. Renal manifestations range from asymptomatic proteinuria to early-onset steroid-resistant nephrotic syndrome. It was originally described in 1968 by Galloway and Mowat in two siblings with the triad of congenital nephrotic syndrome, microcephaly, and hiatus hernia [1]. The majority of patients usually die within a few years of the onset of the disease, and the commonest causes of death are nephrotic syndrome or seizures. Steroid-resistant nephrotic syndrome, a common renal manifestation in GAMOS, is associated with poor prognosis and often progresses to end-stage renal disease [1]. The estimated prevalence is less than 1/1,000,000 [2]. Homozygous mutations in the WD repeat domain 73 (WDR73) were first implicated in patients with GAMOS in 2014 [3]. We report a rare case of an adult who presented with only proteinuria and was found to have GAMOS with a deletion of the WDR73 gene.
Case presentation
A 64-year-old man with a history of recurrent deep vein thrombosis on anticoagulation, intermittent atrial fibrillation, ulcerative colitis, apical hypertrophic cardiomyopathy, hypertension, and biopsy-proven membranous nephropathy (MN) presented with worsening urinary frequency associated with frothing despite being compliant with all his medications for his previously diagnosed MN. The patient’s initial home medications included sulfasalazine 1 g three times daily, folic acid 1 mg daily, apixaban 2.5 mg twice daily, rosuvastatin 20 mg daily, famotidine 20 mg daily, tacrolimus 5 mg twice daily, prednisolone 5 mg daily, metoprolol succinate 25 mg daily, and enalapril 5 mg daily. The patient’s initial kidney biopsy, which confirmed MN, was done in Barbados. The patient denied smoking, alcohol, or illicit drug use but had a significant family history of cancer. The patient’s mother and sister both had breast cancer, and the mother also had colon cancer. He had inguinal hernia repair at 5 and 63 years, respectively. He also had a lipoma removed.
The patient's initial physical examination and vital signs were temperature 97.5°F, heart rate 79 bpm, respiratory rate 16/minute, saturation on room air 100%, blood pressure 132/66 mmHg, and BMI 25.9 kg/m^2^. The patient was euvolemic, not jaundiced, and not pale, and had no pedal edema. The patient’s chest was clinically clear, and abdominal examination was unremarkable. He was conscious, alert, and oriented in terms of time, place, and person. Table 1 summarizes the patient’s extensive laboratory workup, which was largely within normal limits except for elevated proteinuria with a urine-protein-to-creatinine ratio of 2,800 mg/g.
Imaging
The kidney ultrasound is represented in Figure 1. It showed increased echogenicity but normal parenchyma thickness and contour, and no pelvicalyceal dilatation, calculi, cysts, or solid masses in both kidneys. The increased echogenicity and absence of cysts/masses suggest chronic nephropathy.
Kidney ultrasound of our patient.(A) Right kidney of size 10.6 x 5.3 x 5.0 cm with increased corticomedullary differentiation (blue arrow). (B) Left kidney of size 10.6 x 5.3 x 4.9 cm with increased corticomedullary differentiation (blue arrow). (C) Doppler images showing blood flow (blue arrow) of the right kidney. (D) Doppler images showing blood flow (blue arrow) of the left kidney
Kidney biopsy
A repeat kidney biopsy was done to work up the worsening proteinuria. The results of the repeat kidney biopsy are presented in Figure 2. The biopsy consisted of three cores of renal cortex and medulla with 29 glomeruli. Five glomeruli were globally sclerosed. The remaining glomeruli showed irregular capillary wall thickening and spike formation on Jones silver staining, indicative of MN. Deposits were confirmed by immunofluorescence and were positive for IgG and complement 3. Immunofluorescence staining was negative for phospholipase A2 receptor (PLA2R), thrombospondin type 1 domain-containing 7A (THSD7A), and neural epidermal growth factor-like 1 (NELL-1). Electron microscopy revealed subepithelial and intramembranous electron-dense deposits with reactive basement membrane spikes and complete foot process effacement. No mesangial or subendothelial deposits were present.
The patient's biopsy images. (A) Glomerulus showing irregular capillary wall thickening, PAS stain (40×). (B) Glomerulus revealing capillary spikes by Jones silver staining (40×). (C) Granular capillary wall staining for IgG on immunofluorescence (IgG, 40×). (D) Glomerular capillary loops with subepithelial and intramembranous electron-dense deposits and overlying complete foot process effacement (electron microscopy, 2,500×)PAS: periodic acid-Schiff; IgG: immunoglobulin G
The clinical findings of adult-onset proteinuria (urine protein-to-creatinine ratio of 2,800 mg/g), together with kidney biopsy-confirmed MN but negative PLA2R/THSD7A/NELL-1, support the diagnosis of GAMOS-associated MN in the context of WADR73 gene deletion. The patient was started on cyclosporin by a nephrologist in Barbados. We recommended rituximab infusion to decrease the risk of progression of kidney disease, as patients had a poor response to calcineurin inhibitors.
Discussion
We report a rare case of an adult who presented with only proteinuria and was found to have GAMOS with the deletion of the WDR73 gene. GAMOS was originally described in 1968 [1]. Since then, about 60 cases of GAMOS have been reported [4]. The clinical spectrum of GAMOS is vast, and it includes craniofacial dysmorphism, developmental delay, seizure disorder, hypotonia, psychomotor retardation, extremities abnormalities, and diverse renal manifestations [4]. Hiatus hernia was initially part of the diagnostic criteria. However, it is no longer considered a core clinical feature of GAMOS [5]. Our case is a unique form of GAMOS because our patient presented in adulthood and without any neurological symptoms, which is almost always required to make a diagnosis.
Renal manifestations of GAMOS vary from isolated proteinuria to steroid-resistant nephrotic syndrome [3,6,7]. There were no renal abnormalities in a few reported cases [3,6,7]. Clinical renal variability of GAMOS exists even within the same family. For example, two siblings carrying the same pathogenic mutation of WDR73 developed contrasting renal pathologies [3]. One sibling developed nephrotic syndrome at five years, which rapidly led to chronic renal failure and death after a month of diagnosis [7]. The other sibling who had the same pathologic variant of WDR73 had no renal disease even at seven years [3]. Our patient had a pathologic variant of WDR73 but presented with an isolated proteinuria, which also lies in the spectrum of renal disease in GAMOS.
There is genetic heterogeneity of GAMOS. Loss-of-function mutation of the WDR73 gene has been reported in different families with GAMOS [3,6]. Recessive variants in the genes encoding for kinases, endopeptidases, and other small-sized protein complexes, such as 0-sialoglycoprotein endopeptidase (OSGEP), TP53-regulating kinase (TP53RK), TP53RK binding protein, and L-antigen family member 3, have also been identified and linked to GAMOS in different families [7]. A recent in vivo knockdown study of OSGEP and TP53RK led to pathologies in the actin cytoskeleton as well as a reduction in the human podocyte migration rate [7]. These pathological manifestations led to the development of nephrotic syndrome [7]. Our patient had a gene deletion of WDR73, which possibly led to the worsening proteinuria. A case report involving four families showed findings consistent with GAMOS; the affected individuals had a mutation in the Nucleoporin 133 gene [8]. Another case report of four families identified mutations in the gene encoding for the zinc-finger domain of the transcriptional regulator PR/SET domain 15 that resulted in nephrotic syndrome within a few months of life and other features of GAMOS [9]. Our patient's family history was negative for proteinuria or kidney disease.
GAMOS is a rare disease entity with no existing management guidelines. Genetic testing helps in making a diagnosis, but currently, there are no treatment modalities. Management includes managing the comorbidities associated with it. In our patient, an effort was made to reduce proteinuria with medications to reduce the progression of kidney disease. In cases where the disease is detected in childhood, early referrals for genetic counseling and testing, as well as management of comorbidities, are primarily the treatment modalities.
Conclusions
Our case highlights the late onset of GAMOS with nephropathy but without neurologic manifestations. This form of MN with an unknown responsible antigen, with negative staining for PLA2R, THSD7A, and NELL-1, is potentially related to the detected underlying deletion of the WDR73 gene as an atypical presentation of GAMOS. Genetic testing is key in clenching the diagnosis in suspected cases.
The reference list from the paper itself. Each links out to its DOI / PubMed record.
- 1Congenital microcephaly with hiatus hernia and nephrotic syndrome in two sibs J Med Genet Galloway WH Mowat AP 31932151968571364610.1136/jmg.5.4.319PMC 1468664 · doi ↗ · pubmed ↗
- 2Novel homozygous OSGEP gene pathogenic variants in two unrelated patients with Galloway-Mowat syndrome: case report and review of the literature BMC Nephrol Domingo-Gallego A Furlano M Pybus M 1262020193097508910.1186/s 12882-019-1317-y PMC 6458604 · doi ↗ · pubmed ↗
- 3Loss-of-function mutations in WDR 73 are responsible for microcephaly and steroid-resistant nephrotic syndrome: Galloway-Mowat syndrome Am J Hum Genet Colin E Huynh Cong E Mollet G 6376489520142546628310.1016/j.ajhg.2014.10.011PMC 4259970 · doi ↗ · pubmed ↗
- 4Galloway-Mowat syndrome in Taiwan: OSGEP mutation and unique clinical phenotype Orphanet J Rare Dis Lin PY Tseng MH Zenker M 2261320183055865510.1186/s 13023-018-0961-9PMC 6296068 · doi ↗ · pubmed ↗
- 5Galloway-Mowat syndrome of abnormal gyral patterns and glomerulopathy Am J Med Genet Cooperstone BG Friedman A Kaplan BS 250254471993821391410.1002/ajmg.1320470221 · doi ↗ · pubmed ↗
- 6Extending the mutation spectrum for Galloway-Mowat syndrome to include homozygous missense mutations in the WDR 73 gene Am J Med Genet A Rosti RO Dikoglu E Zaki MS 992998170 A 20162700191210.1002/ajmg.a.37533 PMC 5011457 · doi ↗ · pubmed ↗
- 7Mutations in KEOPS-complex genes cause nephrotic syndrome with primary microcephaly Nat Genet Braun DA Rao J Mollet G 152915384920172880582810.1038/ng.3933 PMC 5819591 · doi ↗ · pubmed ↗
- 8Correction Ann Neurol 4624638520193081785710.1002/ana.25427 · doi ↗ · pubmed ↗